An ESO task force met to discuss the potential for “biosimilar” medical products in oncology, analyzing facts and debunking some myths.
 Some of the most used biologic drugs in oncology are going off patent in the near future, and this will give pharmaceutical companies the opportunity to create “biosimilar” drugs. These copies of the originator will potentially increase the access to effective therapies against cancer, hopefully leading to a sustainable cancer care, but many yet unanswered questions might make it difficult for biosimilars to be easily integrated into clinical practice. For this reason, the European School of Oncology created a Task Force to discuss challenges and opportunities related to biosimilars in oncology that will come up in the near future with a position paper.
Some of the most used biologic drugs in oncology are going off patent in the near future, and this will give pharmaceutical companies the opportunity to create “biosimilar” drugs. These copies of the originator will potentially increase the access to effective therapies against cancer, hopefully leading to a sustainable cancer care, but many yet unanswered questions might make it difficult for biosimilars to be easily integrated into clinical practice. For this reason, the European School of Oncology created a Task Force to discuss challenges and opportunities related to biosimilars in oncology that will come up in the near future with a position paper.
What is a biosimilar?
It might seem we are reliving the same old story: years ago, loss of exclusivity of conventional drugs led to the so-called “generic” medicines, very common on the market nowdays. But the paradigm accepted for conventional drugs and generics can not be directly applied to biological medicines and biosimilars, because there are big differences between the two classes of drugs. As a matter of fact, biological medicines are made by – or derived from – a biological source, are generally large and complex molecules, presenting many possible modifications due to the specific living system they are produced by and, last but not least, are immunogenic.
«There is no scientific definition of a biosimilar. It is a regulatory designation that can be different for different agency like EMA or FDA» pointed out Mario Dicato, from Centre Hospitalier de Luxembourg and member of the ESO Task Force on Biosimilars.
«We could also say that a biologic drug is itself a biosimilar of a natural protein» Huub Schellekens, from Utrech University in The Netherlands, added, also highlighting the existing differences between batches of the same biologic drug.
Taking into account all these cogent arguments, companies developing biosimilars must prove «high similarity» of the new molecule to the reference product with approaches that often require multiple iterations of process change and physicochemical characterization. Even a slight alteration in manufacturing of biologics, in fact, can lead to clinically relevant changes.
It’s not just a matter of price
Why do we need biosimilars? Price is one of the forces driving the development of biosimilar drugs: «The place of biologics is increasing in oncology treatments with an estimation of >100 billion USD expenditure for these drugs worldwile today» said Joseph Gligorov, from Sorbonne Université in Paris. Considering that biosimilars are expected to be priced approximately 20% to 30% lower than their reference biologic, they have the potential to significantly lower healthcare costs. This would expand the access to potentially life-saving drugs, especially in developed countries, whereas in the developing countries – where even “basic” drugs can be hard to find – access to biosimilars is not a priority in cancer care. Saving money is important, but it seems it’s not enough to convince physicians to trust biosimilars and to include them in their clinical practice.
«There are a lot of myths on biosimilars still to be debunked» Giuseppe Curigliano, from the European Institute of Oncology in Milan, said. Among them, the belief that «cheaper» means «less effective», that confirmatory trials are too small to detect clinically meaningfull differences, that pharmacovigilance will be poor without different non-proprietary names. Moreover, the willingness to accept a biosimilar also depends on the function of the original molecule. «An oncologist will be more cautious in prescribing a biosimilar in a curative setting than in a non-curative one» said Dirk Arnold from Instituto CUF de Oncologia, Lisboa, Portugal.
Biosimilars in oncology clinical practice
Three of the biologic molecules most commonly used in oncology have already lost or are going to lose exclusivity on EU and US market: Avastin (bevacizumab), Rituxan (rituximab) and Herceptin (trastuzumab). Several companies are now focusing on development programs to be ready to market these biosimilar drugs as soon as the originators will be declared off-patent. One phase I pharmacokinetic (PK) study was designed comparing PF‑06439535 (a potential biosimilar) with bevacizumab from US and from EU; another testing pharmacokinetic and Safety of CT-P10, a biosimilar candidate to the rituximab reference product and so on, with at least 5 companies «on track» for launch at the time of the EU/US loss of exclusivity for trastuzumab.
From a regulatory point of view, for the approval of a biosimilar phase III clinical trials are not mandatory. «The aim of clinical trials with biosimilar is to show equivalence, not patient benefit» Curigliano explained, pointing out that there is an open debate about the end point of such studies. EMA guidelines identify Overall Response Rate (ORR) as a sufficiently sensitive endpoint for clinical trials of biosimilars antibodies even if very often ORR does not correlate with survival. «The candidate biosimilar should be indistiguishable from the reference product concerning physical-chemical characteristics and in vitro biological activity. Moreover it is important to verify immunogenicity and potency of the new molecule» Schellekens explained, suggesting that biosimilars have now comparable quality with respect to the reference product.
Extrapolation and automatic substitution: how safe?
The ESO task force meeting also focused on extrapolation and automatic substitution, hot topics within the debate on biosimilars. Extrapolation is the approval of a biosimilar for use in an indication held by the reference product, not directly studied in a comparative clinical trial with the biosimilar. As the EMA states «the primary rationale for data extrapolation is to avoid unnecessary studies in the target population for ethical reasons, for efficiency and to allocate resources to areas where studies are more needed».
«Extrapolation is something we daily do in clinical practice, when treating patients who are different from the population included in clinical trials» said Curigliano. «Extrapolation of an indication – when scientifically justified – is part of the biosimilar concept of making biological therapies more broadly accessible within a tailored development program» he added.
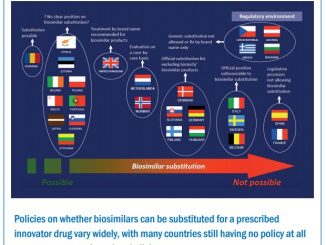
Things change when it comes to automatic substitution, the substitution by a pharmacist without the physician’s consent. Although allowed for generic drugs, there is no scientific basis supporting automatic substitution with biosimilars, and regulatory decisions on the topic are left to individual countries. «These concerns make pharmacovigilance projects after biosimilar approval even more important» Curigliano concluded.