



Since the concept of personalised cancer therapies first emerged, the picture has become so much more complex and challenging.
 “If it were not for the great variability between individuals, medicine might as well be a science, not an art.” Sir William Osler (1892)
“If it were not for the great variability between individuals, medicine might as well be a science, not an art.” Sir William Osler (1892)
The whole concept of personalised medicine is not really new. We’ve been treating patients in a personalised manner for many years; the change is in our ability to understand what we are doing and how we deliver personalised medicine, hopefully leading to improved outcomes.
We are now able to characterise and study each patient and their tumour in a breadth and depth not previously possible, which allows us to be much more precise in the way we manage the individual. I want to change the mantra of personalised medicine – ‘the right dose of the right drug for the right indication for the right patient at the right time’ – to add ‘the first time’. It’s become clearer that the first time we get to challenge the tumour with therapy is the most important time in determining the patient’s outcome, so one of the key goals is give the right treatment first.
In the past, we treated cancer patients with relatively blunt instruments – chemotherapy, radiation therapy – that target primarily the proliferative rate of the tumour. We can now begin to characterise tumours in sufficient depth to identify what drives the tumour and then to target that in a way that capitalises on the changes in the tumour. Normal cells are incredibly robust. In contrast, cancer cells are genomically unstable and have many aberrations, which in many cases render the cancer cell less robust than normal cells in the body. If we can understand these dependencies it should be possible to define approaches that more selectively target and kill tumour cells.
It’s an incredible time. With new technologies and approaches we finally have the ability to let the patient teach us what is important. We have what we hope is a ‘perfect storm’ of two events coming together: the ability to characterise the patient and the tumour on the one hand, and an incredible repertoire of drugs able to capitalise on the genetic changes present in the patient’s tumour on the other. There are almost 1000 different drugs in, or about to enter, clinical trials that target particular underlying events in tumours, including more than 100 in breast cancer alone.
Using response prediction biomarkers
The most effective targeted agents are linked to response prediction biomarkers (see table below). With these, we are seeing remarkable responses in patients in a range of different cancers. The good news is that there are more and more of these. The bad news is that, in many cases, it takes far too long from when we identify underlying abnormalities to when we move a drug into the clinic.
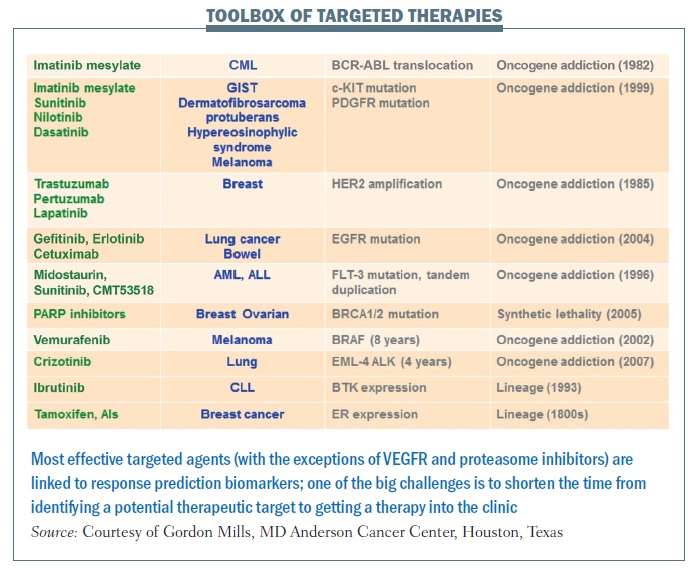 Crizotinib represents what we hope will be the new approach. It took less than four years from identification of a particular abnormality in lung cancer – EML-4 ALK fusion gene – to a drug being shown to be effective in clinical trials and approved for use in this disease, for which we had no other therapy option that worked. Crizotinib was being developed for a completely independent reason. However, it was known to target ALK, and because it was available on the shelf, ready to go, it was very easy to link testing for the EML-4 ALK aberration that occurs in a subpopulation of lung cancer to treating patients with a drug specifically targeting that therapeutic liability.
Crizotinib represents what we hope will be the new approach. It took less than four years from identification of a particular abnormality in lung cancer – EML-4 ALK fusion gene – to a drug being shown to be effective in clinical trials and approved for use in this disease, for which we had no other therapy option that worked. Crizotinib was being developed for a completely independent reason. However, it was known to target ALK, and because it was available on the shelf, ready to go, it was very easy to link testing for the EML-4 ALK aberration that occurs in a subpopulation of lung cancer to treating patients with a drug specifically targeting that therapeutic liability.
In the past, our drug development pipeline has been full of failures. The success rate from phase I to approval in the US in cancer drugs is around 5%. We clearly need to change the way we are doing things. One of the key steps in that process is linking biomarkers that can be used to identify patients likely to benefit to the incredible toolbox of targeted agents. Hopefully, we are entering an era where we can do this much more efficiently and get effective drugs to our patients. For BCR-ABL, identification through to an approved drug took over 40 years. erbB2 inhibition took 13 years, and evaluation of PARP inhibition is still ongoing for BRCA1 and BRCA2 mutation carriers. But for BRAF, identification of the abnormality to an effective targeted therapy took 8 years and crizotinib for EML-4 ALK took only three to four years.
The MD Anderson Cancer Center approach
The way we run projects at the MD Anderson Cancer Center provides a model for how we might move this personalised approach forward (see figure below). Within five years, for all patients likely to enter clinical trials − totalling 30,000 per year, making this a major challenge and opportunity − we plan to characterise all of the actionable aberrations using multiple platforms looking for anything where we have a potential drug or where we can predict patient outcomes. If there is a targetable aberration, we will direct patients to the standard of care where this is available – for example erbB2 amplification targeted therapy in breast cancer − or to clinical trial cohorts, filling them at a rapid rate and so helping to get more effective drugs to patients. Patients with rare events will be offered ‘n of 1’ trials of therapy (where they are the only trial subject) related to what is going on in their tumour.
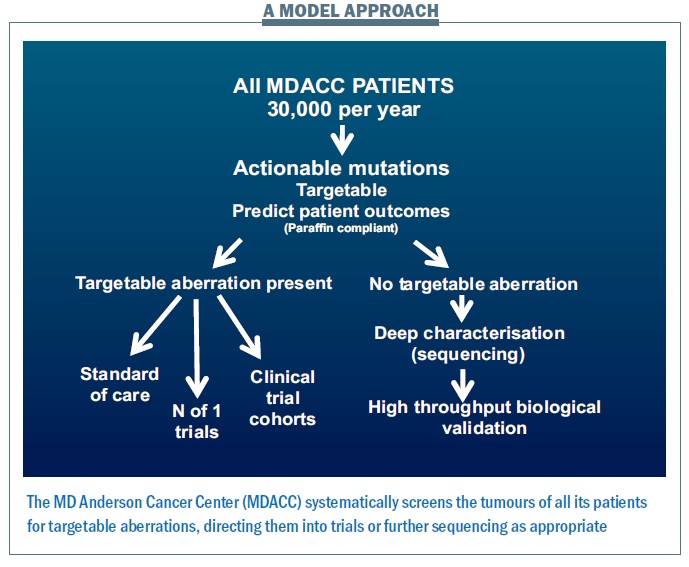 Many patients – more than half – have no targetable aberration present. We then propose to characterise what is going on in much greater depth to try to understand targets that we haven’t previously looked at, and determine whether the patient can benefit from them.
Many patients – more than half – have no targetable aberration present. We then propose to characterise what is going on in much greater depth to try to understand targets that we haven’t previously looked at, and determine whether the patient can benefit from them.
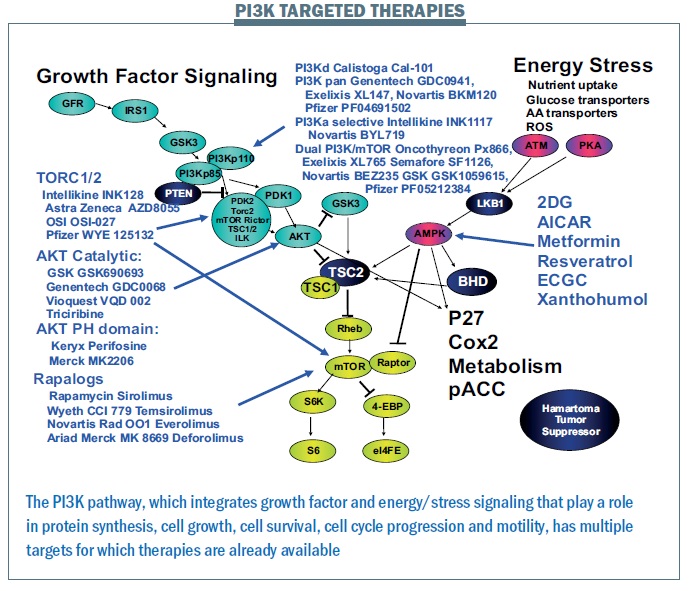
How this can work: the PI3K pathway
The PI3K pathway is proving critically important. We have more mutations in this pathway and more patients that we can target with current therapies than for any other pathway. A wide range of drugs targeting the PI3K pathway are currently in clinical trials (see figure below). Where there is a good toolbox of therapies, the challenge is to identify patients that may benefit.
 The figure below shows data from the MD Anderson phase I programme for patients with mutations in a single gene (PIK3CA) to therapies targeting this pathway. More than 30% of patients are demonstrating benefit, based on RECIST criteria, which is almost unheard of in a phase I programme. Cancers including cervical, ovarian and breast, shown on the right of the waterfall plot, benefited markedly, while other cancers including colorectal, shown on the left-hand side, do not seem to benefit, for reasons that are not yet clear.
The figure below shows data from the MD Anderson phase I programme for patients with mutations in a single gene (PIK3CA) to therapies targeting this pathway. More than 30% of patients are demonstrating benefit, based on RECIST criteria, which is almost unheard of in a phase I programme. Cancers including cervical, ovarian and breast, shown on the right of the waterfall plot, benefited markedly, while other cancers including colorectal, shown on the left-hand side, do not seem to benefit, for reasons that are not yet clear.
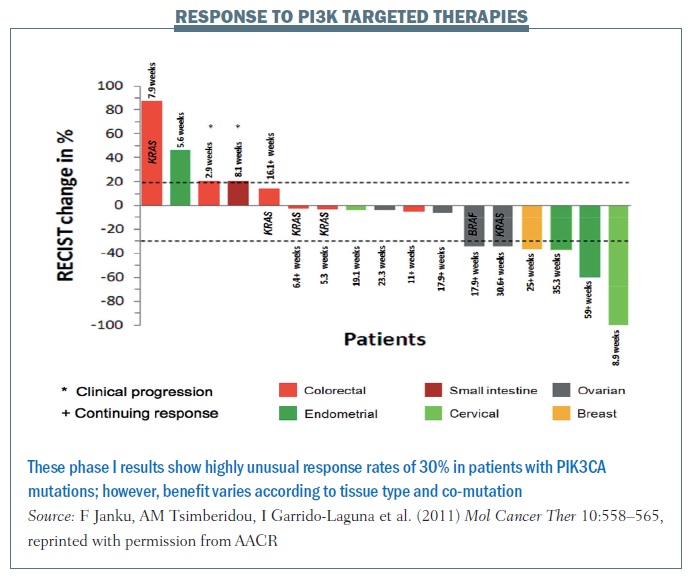 We predicted that co-mutations in the RAS pathway would be markers for resistance but, surprisingly, while RAS mutations in colorectal cancer appeared to confer resistance, two patients with ovarian cancer with mutations in this pathway demonstrated RECIST criteria responses. What does this mean? We believe that linking aberrations to targeted therapy is going to work, but having markers of sensitivity – PIK3CA mutations – is not enough. It will be contextual on the intrinsic gene expression pattern in the patient’s tumour and co-mutations in the tumour.
We predicted that co-mutations in the RAS pathway would be markers for resistance but, surprisingly, while RAS mutations in colorectal cancer appeared to confer resistance, two patients with ovarian cancer with mutations in this pathway demonstrated RECIST criteria responses. What does this mean? We believe that linking aberrations to targeted therapy is going to work, but having markers of sensitivity – PIK3CA mutations – is not enough. It will be contextual on the intrinsic gene expression pattern in the patient’s tumour and co-mutations in the tumour.
Many years ago I proposed that we would have RAS clinics for all patients with RAS aberrations. Looking at our data, and that of many others, we are now thinking of RAS in the context of a specific disease, and managing patients in a disease-oriented programme with an overlay of the genetic aberrations that can be targeted.
Challenges in personalised therapy
The idea of using personalised therapy and being much more effective sounds wonderful. But there are a lot of challenges to be overcome:
How personalised?
Can we really provide a specific therapy for every single patient? This is an anathema to regulatory agencies that want large-scale trials to show improvement in outcomes with a specific drug in a specific disease.
What we are probably going to be doing for a while is precision or stratified medicine: finding homogeneous groups with a particular set of aberrations that are likely to benefit from a particular therapy.
But even that comes with a problem. For example, breast cancer – the most common cancer – has at least eight independent, therapeutically relevant subclasses. One of these is so small that we are unable to mount clinical trials without massive consortia and many years of intervention.
Trials for rare aberrations
Some of these aberrations are quite rare. AKT mutation is one of the key aberrations in the PI3K pathway, but it occurs in about 0.7% of patients going on clinical trials. This means testing thousands of patients to find sufficient patients with this aberration to complete a single trial. Multiplex testing for many different genomic aberrations can direct patients to many different studies, including those for rare aberrations such as AKT mutation.
Small tumours
Obtaining sufficient tissue to test can be challenging with small tumours.
Understanding resistance
Responses tend to be short. We do not understand why resistance emerges in most of the cases, but are attempting to understand this in order to develop rational combinatorial therapy, which will be critical to moving ahead.
We know that positive predictive markers can have only a modest predictive value – only 30−60% of patients with the dominant marker of HER2 amplification benefit from therapy targeting HER2. The rest do not, and we don’t understand why. Unfortunately, when we have a negative predictor such as RAS mutation, it appears dominant over sensitivity.
Side-effects
The pathways that are abnormal in most cancers are the same pathways that function in normal cells, and the question is: can we develop sufficient therapeutic index for targeted therapies to benefit our patients?
Collaboration
How are we going to deal with these major challenges to the field? We are going to need a broad programme – collaborating across many different institutions – where we are able to identify the genomic events driving tumour progression, a repertoire of drugs, biomarkers for individuals likely to benefit, and rational combinatorial therapy.
Pilot project T9
MD Anderson’s T9 pilot project − short for Ten Thousand Tumours, Ten Thousand Tests, Ten Thousand Therapies − is analysing the cancer-causing genetic variations in the tumours of 10,000 patients with advanced cancers that have no standard therapy. Data from the first 1000 tumours analysed in depth showed the frequency of mutations was lower than we had expected: less than 50% of patients had an actionable mutation. There were lower than expected numbers for many of the actionable mutations, which will be important in the design of clinical trials, as it will be necessary to test many more patients than originally predicted.
A facilitation programme, Clearinghouse, which is run through the Institute for Personalised Cancer Therapy (IPCT), helps the faculty at MD Anderson drive clinical trials. A physician contacts us about any patient who is likely to enter a clinical trial – we now have more than 1400 patients enrolled, recruiting more than 300 patients a month. Tissue is obtained and directed to our CLIA laboratory (CLIA indicates its meets Clinical Laboratory Improvement Amendments standards), where it is tested for more than 400 aberrations that are important as targeted events. This is being expanded to an even broader protocol. We also analyse samples for many more events in a research laboratory, giving us an incredible repertoire, speed and cost advantage to be able to identify potential driver aberrations.
Aberrations are reported back to the faculty, following confirmation in the CLIA laboratory, and they use this information to fill their clinical trials, with umbrella or bucket trials, where we may have five or 10 different therapies for different aberrations, and also to direct patients to n of 1 trials. Decision support is provided by a tumour board, and data capture of mutation frequency and outcomes determines that this is truly benefiting patients.
A single patient can change the way we manage cancer
In any clinical trial 5−10% of patients demonstrate remarkable responses, and these patients can teach us important lessons. For example, we started a trial some time ago with sorafenib, meant to target BRAF, which was known to be important in melanoma. One patient responded dramatically. After two months, their melanoma had disappeared completely and has not returned 11 years later. However, we characterised BRAF and found absolutely nothing going on. Looking more deeply, we found a causal mutation in KIT, another actionable oncogene. Based on this plus other data, we now test all patients with acral, mucosal and chronic skin-damaged melanoma for KIT mutation and direct them to KIT-targeted therapy (Nature Clin Practice Oncol 2008, 5:737−740). This accounts for about 30% of acral, mucosal and chronic skin-damaged melanoma. This was a change in practice driven largely by a marked response in one patient, and letting that patient teach us what was important.
Deep molecular characterisation of each of a patient’s aberrations is needed to determine which are drivers and what is the best therapy to target these drivers. This whole idea of deep characterisation of every patient with an underlying mutation or response, to try to determine the best approach, is now emerging as the standard at our institution.
Intratumoural heterogeneity
There is marked heterogeneity in many patient tumours, with intratumoural heterogeneity within a single tumour and marked heterogeneity between primary tumours and metastases. These can be pre-existing, in the primary tumour that seeds the metastases, or due to further evolution after the metastasis has occurred.
How are we going to manage this complex problem of intratumoural heterogeneity? It is important to do multiple biopsies – trying to capture both spatial and time-dependent heterogeneity. Currently, we believe our best approach is to treat the dominant mutation we find. If the patient benefits and then recurs, we need to re-biopsy to see what has changed and what is now the dominant clone that we should treat. For example, in breast cancer we re-biopsy patients when they recur to inform us of the best therapy.
The next step is going to be to move from biopsy to looking at circulating DNA and circulating tumour cells. From our early data we believe they will reflect what is going on in all metastatic sites in the body, giving us a much better way to determine what is the best next treatment for the patient. In our preliminary studies on this, when we know what we are looking for in PIK3CA studies, we have 80–90% concordance between what we find in the blood and what we find in the tumour.
Once we have detected an aberration is a driver, what do we do about it? A recent study showed that not all KIT mutations are equal. About 16% of patients who had abnormalities in KIT had clear responses. However, looking much more carefully showed they fell into two groups. There were those where there was evidence that it was a driver mutation and altering the tumour’s behaviour – recurrent mutations that were functionally important. This study identified KIT mutations K642E and L576P in this group – 40% of patients in this population responded, which included all of the responders to imatinib. The other group comprised mutations seen only once, which were not drivers and did not signal sensitivity to the drug. The lesson from this is that it’s important to know whether a gene is mutated, but it’s also critically important to know whether the mutation drives the behaviour of the tumour, rendering it sensitive to therapy.
Looking to the future, we will need to carry out multiple biopsies, characterising the primary tumour and metastases in sufficient depth to identify subclones before carrying out a trial of therapy targeting the dominant subclone against which we have active drugs. Hopefully the patient will respond and sometimes they will be cured. If they recur, we need to re-biopsy and determine what has changed and what is now the current driver to guide a new round of therapy. If we can convert cancer into a chronic disease with relatively benign therapies we should greatly improve morbidity and mortality for our patients.


