Anna Wagstaff looks at changes in the business, regulatory and science arenas that could open the way to delivering better cancer treatments, faster and cheaper.
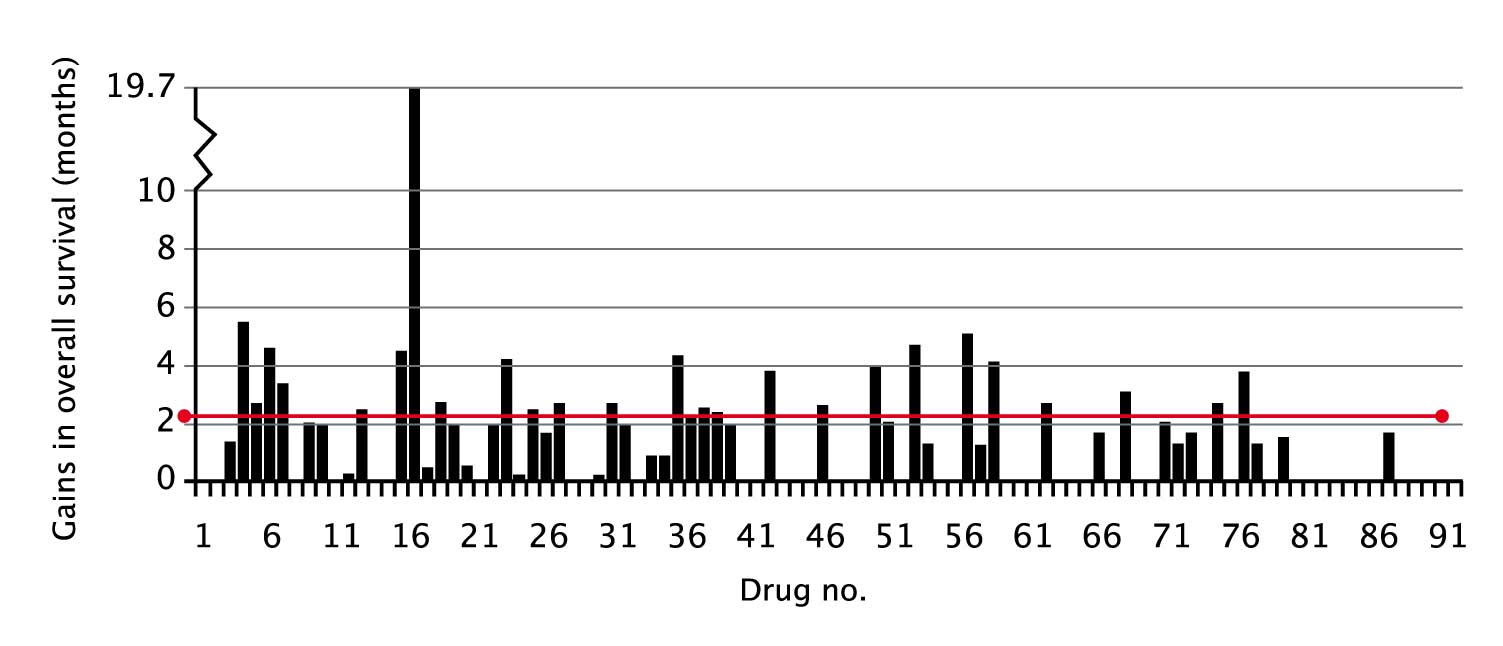
Of 91 new therapies approved for solid tumours between 2002 and 2016, the median reported gain in overall survival was 2.2 months.
Cancer drugs costs are rising five times faster than any other class of medicine.
Eight cancer drugs approved in 2015 have an annual cost of more than $100,000 per patient.
Forbes analysis of average profit margins by sector consistently shows health technology top-ping the league table.
The statistics cited above don’t tell the whole story about the value of these drugs to patients, the costs of developing them, the issues around capturing overall survival data, or the risk and attrition rate involved in drug development.
They do, nonetheless, give grounds for questioning whether the current big pharma business model is the most efficient way to develop drugs in the era of personalised medicines.
Four leading figures in academic drug development addressed this question in an article in Cell published on February 9, which was widely covered in the mass media, including an editorial in the UK newspaper, The Times.
Under the title, ‘How much longer will we put up with $100,000 cancer drugs?’, the authors, from top centres in the US, the UK and the Netherlands, called for “the formation of new relationships between academic drug discovery centers and commercial partners, which can accelerate the development of truly transformative drugs at sustainable prices.”
Speaking to Cancer World, lead author Paul Workman argues that efforts to speed the translation of new discoveries into products that help patients live longer and better have hit the buffers, because the pharmaceutical industry business model avoids the higher risk, more innovative research.
This will continue, he believes, so long as key payers agree to continue paying high prices for low-risk drugs offering incremental benefits.
“You’ve got the fundamental problem that the big pharma companies have an addiction to the big four- to five-billion-dollar drugs like Lipitor, and they just have to price what they have to replace them and keep the business going,” says Workman, who is Chief Executive of the Institute of Cancer Research (ICR) in London.
Current efforts to encourage greater innovation cannot succeed, he believes, because they fail to address the issue of price and sustainability. The emphasis, he argues, is on promoting public–private partnerships aimed at enabling the academic sector to generate more innovative high-risk ideas, and then also do much of the work to ‘derisk’ them, “so that when industry finally does come in, they don’t have to take so much risk, they have a good idea about the patient population, they know the biomarker, they know that maybe a prototype drug is already available and showing promise.”
The flaw in the strategy, he argues, is that even when the lion’s share of the drug development has already been done for them, “the project can often seem to end up with a conventional, large phase III trial model, and payback to the pharmaceutical companies based on the maximum the market will bear.”
He would like to see a dramatic shake up to ensure new drugs can make it to market at a price that is more sustainable and better reflects the extent of public/philanthropic investment in their discovery and development. And he would like to see more drug development done in an academic setting, which he says is more open to taking risks in search of high pay-offs, and better at conducting “small, smart trials”, cutting costs and development time.
Workman has spent 20 years at the ICR building the largest drug development unit within an academic setting anywhere in the world. Since 2005, the ICR has discovered 20 innovative preclinical drug candidates, and taken nine new drugs into clinical trials – among them abiraterone, approved for advanced prostate cancer.
“A mixed economy would probably evolve. It would be a massive change, and a massive change is required”
However, he emphasises that this is not as simplistic as academia versus commercial enterprise. Indeed, Workman says experience at the ICR backs up criticisms from the industry that much ‘landmark’ academic cancer research published in top journals and from reputable labs cannot be reproduced or is not robust across different models.
Are drug developers aiming too low?
(click to enlarge)
The median reported gain in overall survival of new therapies approved by the FDA between 2002 and 2016 for solid tumours was 2.2 months. The graph shows overall survival gains from 91 consecutive drug approvals, starting with imatinib in GIST (no.1, Feb 2002). Where a bar does not appear, it is because overall survival was not a pre- specified endpoint for this indication.
Source: Courtesy Tito Fojo, Columbia University Medical Center, New York. An earlier version of this graph, alongside data for each drug, was published in T Fojo et al. (2014) JAMA Otolaryngol Head Neck Surg 140:1225–36
Workman was himself scientific founder of two successful biotech companies, one of them acquired by Roche, and he sees the biotech sector as an important source of innovation. He has also spent time working in big pharma, including four years heading AstraZeneca’s Cancer Research Bioscience Section. He has huge regard for the quality of the science and the skills within the industry, but is now convinced that their best efforts will not deliver for cancer patients, because the business strategy is inefficient and leads to high levels of duplication.
“Most companies have moved all, or large parts, of their portfolios, almost like a pendulum, away from small-molecule, molecular targeted agents,” says Workman. “Everybody is doing PD-1 and PD-L1 [antibodies that block immune checkpoints]. Everyone wants those in their own portfolios so they can bundle them with other agents of their own, and so they end up with combinations that will be their own… That is massive duplication, adding to costs and also opportunity cost on the innovation that would have been done if everybody just said: ‘We only need, say, three or four of those, and meanwhile let’s get on and innovate with other targets’.”
That opportunity cost, he adds, includes failing to explore the therapeutic potential for targeting the full range of cancer genes that have been identified. “We still only have 5% of the cancer genome covered, so that means 95% dark matter that is yet to be explored. We have to get a heck of a lot more innovative drugs through, with new mechanisms of action, so we can combine them. Otherwise the cancer cells will just find ways to get around them. For these combinations to be created, we have to get those very novel drugs approved.”
Workman believes it is time to trial disruptive approaches for bringing new cancer drugs to market. When appropriately resourced, in expert centres, he argues, academic scientists can discover drugs and work with clinical colleagues to run clinical trials. However, they cannot, and should not, get into the business of marketing their own drugs. He suggests that new forms of biotech or even generics companies, which are already present in the drug manufacturing business, but can operate at far lower profit margins than big pharma, could offer a possible solution.
Under this model, he explains, a drug discovered in an academic setting would be developed with a combination of research grants and charity and philanthropy, or even some venture capital, and at some point would get licensed to one of these new forms of company, which will then be responsible for taking the drug through to approval. “They may need to recruit in skilful people who are very good at more innovative work, and getting regulatory approval,” says Workman, but he argues that it is “addressable”.
Crucially, price caps would be written into the terms of the licence, to keep prices sustainable.
Workman speculates that, with this model, more traditional pharma companies would have to adapt to compete, some would need to downsize considerably and decrease their duplicational marketing costs. “Some would probably go out of business, and some would find creative solutions that would be competitive, and reduce costs to more sustainable levels. A mixed economy would probably evolve. It’s a real possibility. It would be a massive change, and a massive change is required.”
Where is the steam?
Frustration at the slow speed at which new knowledge and understanding about cancer is translating into effective treatments in the clinic is a theme that has been addressed by many commentators in recent years.
Siddhartha Mukherjee, author of The Emperor of all Maladies, is one of them. Interviewed in Cancer World (Sept–Oct 2012), he referred to the widely used metaphor that science inevitably produces a boil that lets itself out as steam through technology. “But if you are living in the world of cancer, there is a lot of boil, especially from the basic science world, but there is little steam which would make the engine move. … So we have all this knowledge, and the public is asking, and we are all asking: where are the medicines that come out of this knowledge?”
Lack of innovation from the pharmaceutical industry and biotechs was one of the problems he identified. “I’m waiting for good exemplars of this change in which the drug emerges from research performed primarily by biotech or pharma companies. I’ve yet to see that. The reality typically still remains investigator-initiated trials or protocols.”
Signs are emerging, however, that the head of steam is beginning to find a way out in the form of new business models more heavily geared towards innovation.
Boston-based PureTech Health is a good example of one such new model. Its focus is on addressing intractable problems across life sciences by scanning the horizon to identify “breakthrough” science at an early stage, and steering it through its preclinical and clinical development, in partnership with the principal investigator and a team of drug development experts.
Last year, Siddhartha Mukherjee became one of those principal investigators, when PureTech Health launched a new company, VOR, around a core technology licensed in from Mukherjee’s lab at Columbia University, where he is working on developing CAR T-cell therapies in a novel way that could extend their application to tackling some of the hardest to treat cancers.
Established in 2005, PureTech Health has a number of products at human proof-of-concept stage, two of which are in pivotal trials to gain market approval. The company has a star-studded top team, including a Nobel Prize winner and many scientists with impressive track records within biotech, pharma and academia. It was floated on the main market of the London Stock Exchange in the summer of 2015.
“What really distinguishes us
is the balance between big academic science
and practical experimental work”
Aleks Filipovic is one of the PureTech Health scientists charged with scanning the horizon for cancer products. She is a practising clinician, who developed monoclonal antibodies against a novel target for invasive breast cancer for her PhD at Imperial College, London, and went on to do a stint at Bristol Myers Squibb as an Associate Medical Director. She clearly enjoys her current job.
“What really distinguishes us from everyone else is the balance between big academic science and practical experimental work. We search for breakthrough academic science which, for example, hasn’t even been published yet, and we will develop the project in collaboration with the scientist.”
The PureTech Health business model relies on ensuring that the early preclinical development is done with the speed and rigour that allows hard-nosed business decisions to be taken quickly, says Filipovic. “We parallel source many experiments, we have weekly calls with our scientists and we do reviews rigorously, with general preclinical development completed more quickly, bringing us to the point where we can apply for a phase I trial. It cuts the development time greatly and it gives us reassurance, because we understand the science in depth. We ask ourselves the hard questions.”
From an industry perspective, one of the key things about this model is that it aims to build sustainable ‘product platforms’, rather than one-off bubbles that collapse once a product makes it (or fails to make it) to market. “We like a platform-based approach, so that this particular technology can give us a lead product, but there is a pipeline behind that can be developed as the programme matures.”
This template, she says, is already working in some of their more mature platforms.
Third Rock, another Boston based company, established in 2006, professes a similar strategy, claiming to “discover, launch and build great companies based on bold ideas that meet at the intersection of science, strategy, business and medicine.” One subsidiary, Igenica Biotherapeutics, is developing novel antibodies and antibody drug conjugates for treating cancer, while another, Constellation Pharmaceuticals, specialises in epigenetics and chromatin biology, and is looking to develop novel cancer therapeutics that target transcriptional pathways and acquired dependencies in tumour cells.
Will these innovation-led companies be the model of the future? “I hope so”, says Filipovic, “because this is where science and the clinic meet in the most meaningful way to really address the unmet medical needs.”
It’s a good model for rapidly taking innovation from academia and getting effective new therapies to market, agrees Workman. What it won’t do, he suspects, is make these new therapies any more affordable.
Not-for-profit innovation
One solution to the affordability problem could be offered by the increasing investment in drug development that is being financed through philanthropic foundations and charities.
Much of this is quite fragmented and focused on particular cancer types – often driven by patient advocacy groups. Sarcoma UK, for instance, recently announced it had raised more than £1 million for research, which has financed work on new treatments, for instance, for chordomas and advanced sarcomas.
But there have also been moves to consolidate cancer research funds into sizeable investment companies that can emulate the business model adopted by PureTech Health and Third Rock, but in a not-for-profit setting.
Syncona, for instance, was set up in 2013 by Wellcome, the world’s largest medical research charity, with the aim of creating an expert team to establish and operate healthcare companies built around innovative life science technology.
By May 2016, one of its start-ups, Blue Earth Diagnostics, had achieved its first US product licence – for an injected imaging agent which shows the parts of the body where prostate cancer has recurred after treatment.
Syncona’s portfolio includes Autolus, focused on developing novel CAR-T cell therapies, and Achilles Therapeutics, focused on therapies developed around the work of Charles Swanton, at the UK’s Francis Crick Institute, that target tumour neo-antigens that originated from trunk mutations (see also Cutting Edge p 14).
At the end of last year, the money available to fund these sorts of start- ups took a quantum leap with the announcement that Syncona, together with the sizable investment fund from Cancer Research UK, will be absorbed into BACIT – the Battle Against Cancer Investment Trust – to create a £1bn fund that aims to become a “national champion of life science investing”.
GBM AGILE: a model of efficiency
GBM AGILE is an example of an innovative trial design being used to speed up progress by testing multiple therapies across a range of subgroups – in this case patients with the highly aggressive brain tumour glioblastoma multiforme.
Timothy Cloughesy, Director of the Neuro-Oncology Program at the University of California, Los Angeles, describes this as a ‘platform’ trial, because it uses a single infrastructure, “to ensure harmonisation with regard to imaging, tissue acquisition, and how the clinical trial data run through,” and also a single control arm where patients receive the current standard of care.
Once the platform is established, any number of therapies can be tested in the different patient subgroups, in what is envisaged as a “continual process”. Patients are recruited into a control arm, or one of several experimental arms. Data from the different arms are then regularly assessed, so that trial arms that are performing badly can be terminated, new arms can be introduced, and if certain treatments appear to work particularly well for certain patient subtypes, patients with that subtype will be more likely to be randomised to those treatment arms as the trial goes forward.
While the experimental arms come and go, the control arm remains constant and continues to accrue patients, which gives the study precision and statistical confidence.
AGILE will start with three different populations in each trial arm: patients whose disease has recurred following standard treatment, and previously untreated patients, who will be separated into those with and without MGMT methylation. The hope is that, as the trial progresses, more biomarkers will be identified that predict for greater response to particular treatments.
There is almost no limit to the range of therapies that can be trialled using this platform, says Cloughesy: “small molecules and antibodies, agents that affect the micro-environment of the tumour, viruses, vaccines, checkpoint inhibitors, standard chemotherapies, and even different ways of delivering radiation”.
What makes the trial uniquely efficient is that the GBM AGILE team unanimously agreed that agents that show strong evidence of efficacy for a given patient group should not have to prove themselves from scratch in a phase III trial – one of the big frustrations mentioned by Paul Workman. The FDA agreed that GBM AGILE could continue as a second stage within the AGILE framework right through to registration, which will save time and money.
So can the big pharma model that thrived in the era of block buster medicines still survive in the more fragmented and complex era of personalised medicine?
Anne White, Vice President, Next Generation Development & Project Management, and Christopher Slapak, Vice President, Oncology Early Phase Clinical Research at Eli Lilly, accept that the personalised medicine paradigm, and the sheer speed at which science is progressing, do pose a challenge. But they argue that the industry has responded by learning to work much more efficiently, through increased collaboration between companies and by partnering with academic bodies.
Lilly, which is one of the top 10 pharma companies for oncology, was given a prominent mention in a 2012 Drug Discovery Today review for its involvement in innovative precompetitive public–private partner-ships and open innovation (vol 17, pp 1088–102).
White singles out TransCelerate, a non-profit organisation set up in 2012 to help “simplify and accelerate the research and development of innovative new therapies”, as a prime example of the new more collaborative approach. Today, she says, Lilly works alongside 17 members of TransCelerate – including almost all the big names in cancer drugs – on a range of initiatives.
She mentions, as an example, efforts to coordinate training of staff at clinical research sites, and to harmonise trial protocol formats. “Right now every pharma has its own protocol templates. Now we are standardising across the industry for our academic partners to read our protocols, and consistently find where the drug information or the dosing is, or the eligibility criteria. That is a really nice example of streamlining to improve efficiency,” says White.
An agreement involving some of the TransCelerate members also facilitates sharing of clinical trial material, “If another company wants access to our medicines, if they are part of this Comparator Network we share materials and we expect the same. That has helped advance science quite a huge amount. These exchanges have potential to speed trials and reduce clinical trial costs and complexity, reduce the risk of unblinding and improve patient safety by ensuring that comparators are used as intended.”
The company is also embracing big data, as a decade of efforts to force the industry and academia to open up access to all trial data are beginning to pay off. To this end, Lilly has invested in advanced analytics: “If we are designing for instance a new trial, we always looked at all the applicable data we had, but now we are able to do Bayesian statistics on a broader set of data, which potentially helps you better predict, for instance, what size the study needs to be to show the difference that you desire.”
“It’s all about finding ways to do things
faster and better, and it’s a process of evolution”
Trial designs are also changing in an effort to identify a defined target patient population as quickly as possible, says White. “We used to start a new trial to ask every question: ‘Can you combine it with this agent?’ ‘Is it effective in this tumour type?’ Now we often design from the get-go multi-arm studies that can roll directly from part A to part B, without having to go through the difficulty of starting a new trial.”
They’ve also managed to cut the total time to enrol to combined trial phases by around a year, through working closely with advocacy groups and clinicians, says White. Trial protocols are now routinely given a “dry run” by clinicians at the testing sites, using theoretical patients, to help ensure any issues that could deter people from joining or sticking with the trial are identified and ironed out before the trial starts.
It’s all about finding ways to do things faster and better, and it’s a process of evolution, she says, a continuous process to “learn, confirm and make adjustments as we go along.”
Responding to the charge that pharma are risk averse and swing behind the latest ‘big thing’, White argues that companies need a mix of high-risk and lower-risk products in the pipeline to be sustainable, but that addressing unmet medical need is an important criterion. “A portion of our portfolio very much says we want to be first. And with that comes more risk. You need a portfolio that has a mix of high-risk and ones that you believe you will improve on what is already out there.”
Slapak points out that, while the company does have its own PD-L1 antibody in the pipeline, it has also just delivered the first new therapy for first-line treatment of advanced soft tissue sarcoma for more than 40 years.
Olaratumab, a novel PDGFRα anti-body, was approved by the FDA last year on the basis of data from the phase II portion of the pivotal phase 2 trial showing almost 12 months benefit in overall survival, almost doubling the survival using the erstwhile standard of care.
He adds that partnerships with academic institutions, such as the Harvard-affiliated Dana-Farber Cancer Institute, help them stay abreast of the science and “tap into some of the best innovation out there”.
In the past, he says, they used to just design a protocol and hand it to an institution and say: “Please find patients”. “Collaborations are now also about the best path forward for the molecule, the right patients etc. We propose areas of work and we are open to what they think. We listen to what they say and we solicit proposals from their investigators and we review them.”
From the company’s point of view, says White, learning to do things faster and better has also been driven by an effort to offset costs. “We face increasing drug regulations and expectations by regulators and by payers and reimbursement groups, she says, adding that rising prices across healthcare also contribute to pushing up the company’s drug development costs, as pharmaceutical companies reimburse many of the procedures that patients receive as part of the trial, “so we incur that cost as well.” On top of that, of course, comes the higher costs associated with developing biologicals compared with small molecules.
Change is coming
Richard Barker is Director of the UK’s Centre for the Advancement of Sustainable Medical Innovation (CASMI), an independent, non-profit body uniting Oxford University and UCL (University College London) with a mission “to create new, sustainable models of the medical innovation process to translate advances in basic research into patient benefit more quickly and effectively.”
He comments that many of the new ways of working described by White are broad trends across the pharmaceutical industry. “There is no doubt that both academic efforts and biotech efforts are tremendously important in the pipeline of cancer drugs, and you are beginning to see larger companies reaching beyond the biotechs to make relationships with networks of academics. Across the US a number of different cancer centres are coming together to work with pharma companies to find targets and potential early leads on new drugs.”
But he also points to changes in clinical trials and regulatory practices that he believes could make it much easier for smaller companies and philanthropic organisations to bring new drugs to market.
CASMI has played an important role in discussions around proposals for using ‘adaptive pathways’ to speed up regulatory processes and health technology appraisal for certain categories of drugs, eg where there is demonstrated unmet need and early data suggests a positive risk–benefit profile.
Adaptive pathways could offer an alternative to large and lengthy phase III ‘pivotal’ trials, which require the sort of money to which only major pharmaceutical companies have access. These pathways would rely instead on a development plan tailored to the drug in question, which would provide enough information on risk versus benefit to enable an early decision on conditional approval for use in a specific patient population, followed by monitoring of the drug’s effectiveness and safety in a real-world setting.
This adaptive pathways model was piloted last year, and the European regulator, the EMA, says it is now committed to “further explor[ing] the adaptive pathways concept as an approach to bringing promising medicines to patients with an unmet need in a timely manner.”
Barker also points to the potentially disruptive impact of innovative types of trial, such as basket trials – where a targeted drug is tested across a range of cancers in patients who test positive for the relevant target – and umbrella trials – which test a range of drugs in patients and patient subgroups in a single disease.
He mentions as an example of the latter the I-SPY trials, which started in 2002 and have focused on trying a range of drugs in eight molecular subtypes of patients with stage 2 or 3 breast cancer, and the Lung Matrix trial, looking at different treatments in eight subtypes of patients with non-small-cell lung cancer.
“I think the general feeling in the industry is that this is the direction of the future, where we have enough knowledge of those mutational patterns to combine forces across companies and try to home in on the populations most likely to respond to each drug,” says Barker.
Running trials in this way is hugely more efficient than each company running its own trial on every subgroup. That means it’s a good deal for companies that might otherwise have run their own separate trial, and by lowering development costs it could also help open the market to smaller commercial and philanthropic organisations (see also GBM Agile box).
And as Barker comments, the landscape of drug development is already opening up. He mentions in particular the involvement of patient organisations, which he says “are increasingly investing in research, and sometimes creating their own molecules.”
He suggests that, “if disruption of the current business model is needed, then patient organisations can play a very major role. “If they are funding a trial they can do it with different rules of engagement if they so choose, depending in part on who is coming up with the investment money.”
Could this all add up to the “massive change” that Workman at the ICR is so keen to see? On the basis of this evidence, maybe it could.
Item 1: Improving value for money from public investment in academic drug discovery and development, by using new types of partnership agreements to bring them to markt.
Item 2: New interest – both commercial and philanthropic – in investing in and incubating highly innovative science at an early stage.
Item 3: Changes within the pharmaceutical industry towards greater efficiency through more collaboration within the industry and with academia.
Item 4: Changes in the way drugs are being trialled and in the regulatory processes that bring down costs and speed up results.
Item 5: Increasing involvement of patient and philanthropic organisations in the drug development arena.
There may also be an item 6.
At his pre-inauguration press conference, US President Donald Trump flagged up his intention to introduce national price negotiations for drugs, for the first time in the country’s history. If that happens, it could, for instance, increase the incentive to go for novel drugs that could make a big difference, by reducing the rewards for bringing ‘me too’ drugs to market.
One way or another, it seems change is on the way.
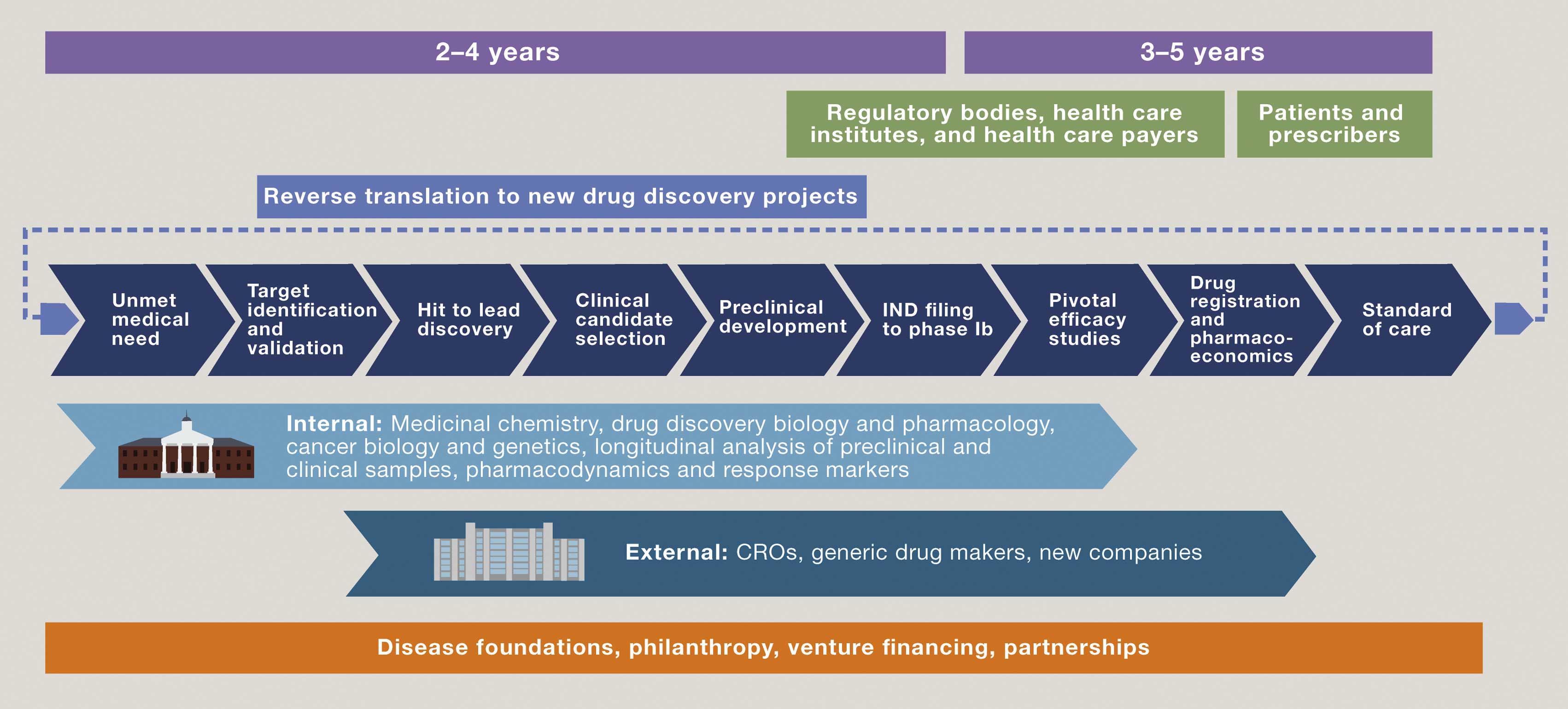
A new paradigm for not-for-profit drug development
(click to enlarge)
Key academic figures in the UK, US, and The Netherlands have issued a call to redefine relationships between academic drug discovery centres and commercial partners, in order to accelerate the development of “truly transformative drugs at sustainable prices”. In an article published in Cell, they argued that, through comprehensive integration of expertise within an academic setting, cancer biologists and geneticists, drug discovery scientists and pharmacologists are able to precisely formulate a ‘Clinical Candidate Profile’ based on tumour subtype(s) and patient population that might best benefit from treatment. Financing can come from a variety of sources, including philanthropic foundations. Not-for-profit entities can retain control right through to commercialisation, by partnering with clinical research organisations (CROs) and generic drug makers or other new forms of company.





Timothy Cloughesy, Director of the Neuro-Oncology Program at the University of California, Los Angeles, describes this as a ‘platform’ trial, because it uses a single infrastructure, “to ensure harmonisation with regard to imaging, tissue acquisition, and how the clinical trial data run through,” and also a single control arm where patients receive the current standard of care.