




When new cancer therapies regularly become available more than half a year earlier in the US than in Europe, or get regulatory approval on one side of the Atlantic but not on the other, patients and clinicians want to know why.
Few issues in healthcare are as emotive as access to new cancer drugs, as they are often seen by patient advocates, politicians and the wider public as lifesaving treatments. In Europe, the agencies in the frontline of recommending drugs for use in national healthcare systems, such as NICE for England and Wales, bear the brunt of criticism for turning down drugs on cost-effectiveness grounds, and for slowness in considering new agents. But in the European Union, no oncology drug can even make it to this stage without marketing authorisation from the European Medicines Agency (EMA).
Since November 2005, all new cancer agents have to be approved centrally for the EU by the EMA, and there has been a growing interest from various players – national medicines agencies, the pharmaceutical industry and patient groups – in its decision-making processes and how they compare with what many consider to be the ‘gold standard’ approval body, the Food and Drug Administration (FDA) in the US.
While new drugs are often available in clinical trials, and existing drugs are sometimes used ‘off label’ in indications for which they have not been approved, widespread use and potentially massive financial returns to drug companies depend on precious marketing authorisations in Europe and North America, and increasingly in Asia. But despite the submission of identical drugs and supporting data from the same clinical trials to both the EMA and FDA, the two regulators can arrive at different authorisation decisions, both initially and when reviewing new data for an already authorised agent. In the past few years, a number of papers and editorials in oncology journals have looked at the reasons for the different decisions, as the answers are not immediately apparent – and even when subject to close scrutiny, authors have not been able to find clear predictors of regulatory outcomes.
But they have detailed differences that can affect clinical practice. When in 2011 Francesco Trotta and Giovanni Tafuri at the Italian Medicines Agency, and colleagues elsewhere in Europe, looked at 100 indications for 42 cancer drugs evaluated by the EMA and FDA between 1995 and 2008, they found that 19 indications were not approved by one of the agencies and 28 had different label wording, ten of which they said had significant clinical meaning (JCO 29:2266–72). Differences noted range from use in treatment, as with Nexavar (sorafenib) – a drug approved for second-line treatment for kidney cancer in the EU but first-line in the US – to a decision that attracted considerable attention, when the FDA withdrew an authorisation for using Avastin (bevacizumab) for advanced breast cancer following new data, while the EMA kept its use in combination with chemotherapy.
“The possibility that the two agencies come up with different decisions about the same drug application may generate confusion both at the level of health professionals and in society at large. We felt this topic deserved a thorough investigation,” says Tafuri. “In particular, the definition of a therapeutic indication is a critical step in regulating medicinal products, and differences in the wording of indications can have a huge impact on clinical practice by including or excluding certain patient populations from the available therapies.”
Most if not all of the differences in authorisations in oncology are not about drugs with clear efficacy benefits compared with risk, but concern agents where there is highly complex detail about narrow therapeutic margins between benefit and harm, so it may not be surprising that different committees of scientific advisors can in turn influence decision makers at the EMA and FDA to come down narrowly on one side or another.
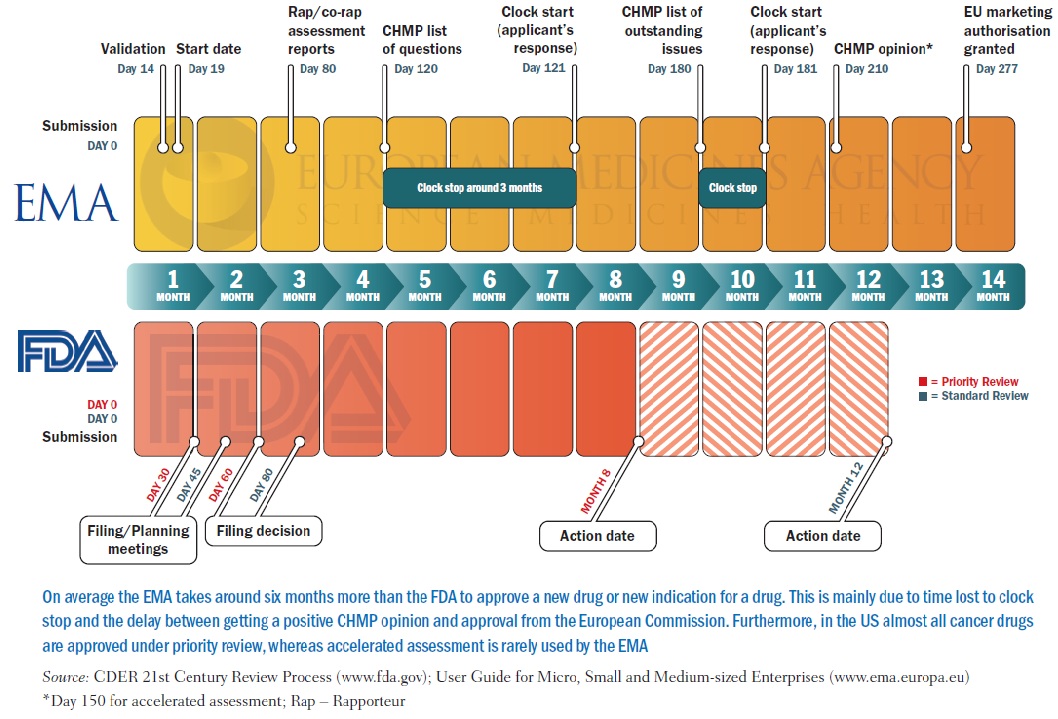
Differences can also arise because of timing and the options open to the regulator, in particular the FDA, which is often the first to receive an application for a drug, and also tends to implement more fast-track and conditional procedures than the EMA (in 2012 the FDA was mandated that it could apply a ‘breakthrough therapy’ designation for serious or life-threatening disease). So new data can also appear during the gap between decisions by the two agencies, and certainly approvals in the EU can take a lot longer than in the US.
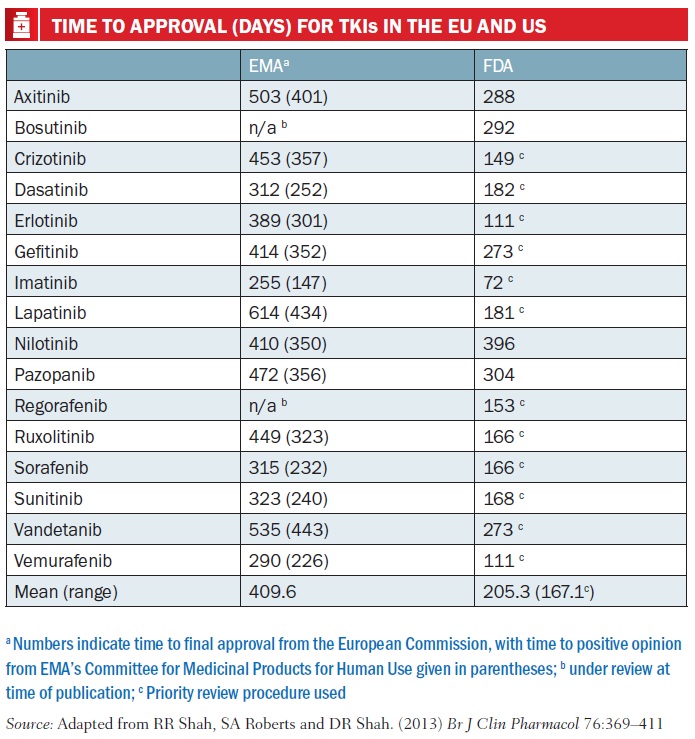
As Rashmi Shah and colleagues report in a review published last September comparing approval of tyrosine kinase inhibitors (TKIs, such as Glivec/imatinib), approval times in the EU were on average twice as long as in the US – 205 days vs 410 days. Most of the delay was due to ‘clock stops’ arising from requests for clarification during the review process, and also the time lapse – about 90 days on average – between a new drug receiving a positive opinion from the EMA’s key body, the Committee for Medicinal Products for Human Use (CHMP) and final approval being granted by the European Commission (Br J Clin Pharmacol 2013, 76:369–411).
The authors also consider that the delay has little impact on public health, as TKIs mostly have only small benefits. They do make suggestions for shortening the EU approval process, such as by using accelerated assessment, “a procedure hardly ever used”, and note too that actually gaining reimbursement for these often very costly drugs at national level is often a source of much longer delay.
Clinical relevance
This is not a static field. The EMA, FDA and others need to develop new processes to cope with a pipeline of new agents, such as immunotherapies, taking into account new science and different ways of quantifying and qualifying the benefit–risk balance. Relations with patient groups, as well as health technology assessment (HTA) agencies are evolving – and the ‘goalposts’ for approving agents with only minor benefit may also be changing. As Markus Hartmann, of European Consulting and Contracting in Oncology, comments, it is just as important to be aware of how each regulatory agency is changing its own approach, rather than focusing purely on how their outcomes compare with those of their main counterparts.
“There is a big discussion about statistical significance versus clinical relevance, and it is now the case that, despite positive clinical trial outcomes, a drug company may not get marketing approval,” he says. “A good example at the EMA is with Tarceva (erlotinib), which was finally approved by the agency for pancreatic cancer after a controversy about its very small but statistically significant gain of 0.3 months in overall survival. But then in 2009 the agency turned down Merck’s Erbitux [cetuximab] for first-line metastatic non-small-cell lung cancer although there was similar significant survival data, from a phase III study including more than 1100 patients, demonstrating an overall survival gain of 1.2 months.”
Hartmann, and colleagues from Merck, have since looked in detail at underlying parameters such as hazard ratios that could be a guide to when the EMA is likely to approve or not approve a new drug, and also when it could gain accelerated approval (Crit Rev Oncology/Hematol 2013, 87:112–121).
Francesco Pignatti, head of the EMA’s section that coordinates marketing authorisations for oncology products, says: “A number of things have changed over the past 18 years or so since I came to the EMA. For example, the first assessment report produced by the Agency in 1999, for docetaxel, was only a few pages long. Now they typically run to hundreds of pages. I don’t think the criteria for assessing oncology products have fundamentally changed, but we try to communicate more transparently the data and justifications for the Agency‘s opinions. This is continuing – we now have a proposal for making public, under certain conditions, the clinical trial data on which authorisations are based.”
“We try to communicate more transparently the data and justifications for the Agency’s opinions”
Transatlantic differences
There are important differences to note with the FDA, he says. One is that it issues investigational status (IND) for new drugs in clinical trials – this is managed by member states in the EU. “A company can do all its development up until seeking authorisation without the EMA being actively involved,” says Pignatti, who adds though that companies do ask for scientific advice during development, such as about clinical studies that have to be submitted, and about requirements for paediatric indications and orphan drugs. The EMA also manages a database of clinical trials.
“Another fundamental difference is after submission for authorisation – the FDA carries out its own analysis of patient-level data to replicate main analyses or to explore possible bias, sensitivity to assumptions and so on. We don’t do that systematically – if we need to explore something, we generally ask the company to submit more details. Some have criticised us, saying that we should do similar in-depth analysis ourselves, but I can’t say that is necessarily better – and if we receive an application after the FDA has done this, the process is partly redundant anyway, at least when replicating analyses. But it is possible we will do more of such analyses in the future, as we do of course receive some drugs for authorisation first.”
There is a lot of collaboration between the agencies under a confidentiality arrangement. The EMA and FDA will give joint advice if requested by a company, and there are monthly teleconferences (also with Health Canada) in a so-called ‘oncology cluster’. “We discuss issues such as ongoing drug application reviews, advice on clinical trial design, and when early approval mechanisms are being considered,” says Pignatti.
One big difference between the EMA and the FDA is that the former is itself an exercise in collaboration. EMA’s CHMP has members from all EU countries and is informed by statutory scientific advisory groups (made up of academic experts and patient representatives). With the expansion of the EU, inevitably it has a much more complex structure than the equivalent review group at the FDA.
As Tafuri explains: “The EMA is based on a network of national regulatory agencies, which has certainly contributed to increasing the level of communication and harmonisation across national agencies. However, even between the EU member states, achieving harmonisation has often proved to be an onerous process, requiring legal referral and arbitration procedures for resolving disharmony.” Tafuri and colleagues have also recently conducted a qualitative interview study concerning the assessment of cancer drugs as well as the reasons for regulatory divergence between the EMA and FDA, which he says will be published soon in Annals of Oncology. “Interestingly, we found that although factors related to the data package of the drug application are the main drivers for regulatory decisions, the influence of factors unrelated to the data, such as the level of interaction with external stakeholders (e.g. pharmaceutical companies or patients), as well as sociocultural and behavioural aspects, play an important role in the drug evaluation process.”
EMA’s Pignatti feels that, while cultural and political factors undoubtedly do play a part, given the diversity in Europe, “just small changes in clinical judgement can make the difference in approving drugs that have very narrow benefit–risk balance.” As he says, weighing up multiple factors such as survival, symptom improvement, response, quality of life, toxicity and more is difficult, and expert judgment still comes into play.
 When opinions diverge
When opinions diverge
Having said that, differences between the EMA and FDA, when they occur, can have a big impact and also result in heated debate. The decision on Avastin in breast cancer is one with major implications, given the prevalence of the disease. Pignatti says that, in his view, although both agencies consider progression-free survival (PFS) as a reasonably likely surrogate endpoint, “we considered that PFS could be a relevant clinical endpoint in its own right – this has been clear in our anticancer guideline for many years. It won’t have the same weight as overall survival, but still some weight.” The FDA, in contrast, when sufficient benefit in overall survival did not materialise in further trials, and given the toxicity profile, took the view that Avastin should be withdrawn for the combination indication (with paclitaxel) for metastatic breast cancer.
But in another case the EMA did not approve Avastin, this time in recurrent glioblastoma, as it was not convinced by phase II trials and the efficacy data that led the FDA to grant accelerated approval. There have been outspoken views on this, with one US proponent having said the data is “unchallengeable”, while European experts said there were too many unanswered questions and that accelerated assessment on uncontrolled trials could set bad examples for drug development.
Both Pignatti and Hartmann comment that there is little evidence that one agency is more conservative than the other, given the diversity in judgements where there is a difference, and that in most cases the decisions reached are the same. Everyone in oncology is grappling with how to assess clinical relevance, says Hartmann, and there will be increasing pressure to introduce new drugs earlier in treatment lines for ethical reasons, but this will make overall survival harder to assess for a certain agent and puts more emphasis on wider quality-of-life benefits and surrogate measures of possible success.
At present, Pignatti says, the FDA does tend to look at PFS as a likely surrogate for overall survival and requires confirmation of this, as in the Avastin breast cancer case, unless the effect on PFS is very large. The EMA, however, takes a slightly different approach. “Our scientific advisory groups, oncologists and patients have said that it is valuable for a patient to delay disease progression and likely worsening of symptoms, and not to have the anxiety of a doctor telling them the tumour is progressing and maybe then having to switch to a less effective therapy. We don’t put a limit in terms of minimum clinically relevant effect size for PFS, as this depends on the balance with the risks.”
Tafuri adds: “It may be time for the oncology community and regulatory agencies to take a hard look at PFS and reflect on whether this can be used as a primary efficacy endpoint in a specific oncology setting.”
“It may be time to take a hard look at whether PFS can be used as a primary efficacy endpoint in a specific setting”
As Hartmann says, this can also be seen as part of a move to incorporate patient groups much more in evaluating what factors are important in the benefit–risk assessment of drugs. The EMA will be adding an appendix on quality of life and patient-reported outcomes to its main guideline, ‘Evaluation of anticancer medicinal products in man’, and last summer the FDA started a series of public workshops on understanding patient needs.
In September, the EMA also held a workshop that explored ways to further involve patients in the benefit–risk assessment of medicines. Currently there are no patient representatives on the main committee (the CHMP), but Pignatti stresses they are on scientific advisory groups and on some committees, such as for pharmacovigilance risk assessment and orphan medicines.
Engaging with HTAs
And all patients and oncologists are concerned about the health technology assessments and cost–benefit evaluations of drugs – because they are often approved only to be turned down by reimbursement and HTA agencies on the grounds of cost-effectiveness. As Olli Tenhunen, an oncology specialist at the Finnish Medicines Agency, comments: “There is more to be done in terms of reimbursement procedures and HTAs. These are not harmonised in Europe, and there seems to be a significant gap between the marketing authorisation and national reimbursement.”
While it is not part of a medicine regulator’s remit to consider cost-effectiveness, the EMA does engage HTA organisations in so-called parallel scientific advice for drug development, and there is ongoing work, including pilots with the European Network for Health Technology Assessment (EUnetHTA), to address concerns that “sponsors are not sufficiently addressing the varying evidence needs of payers and healthcare-guidance and HTA bodies in their medicine-development programmes”.
Hartmann says that the need for them to do so is increasing following legal initiatives such as the Cross-Border Healthcare Directive (which came into force in October 2013, and has an HTA component informed by the EUnetHTA). There is also now a need, he adds, to include a relative effectiveness assessment in risk management plans for drugs, which also also points to such assessments being explained more fully in submissions to the EMA. He notes too that in the US there is now a legal basis for comparative effectiveness research, and the FDA is empowered to enforce the conduct of post-marketing studies and to assess effectiveness data after approval.
 Transparency and trust
Transparency and trust
If the greater emphasis on HTA is one big point, another major move on both sides of the Atlantic is indeed to upgrade the frameworks for benefit–risk assessment for qualitative as well as quantitative approaches. Pignatti confirms this is the case for the EMA. “We are piloting a new template on benefit–risk so that the CHMP can be more explicit about the reasons that matter for the evaluation, and about where there are value judgements,” he says.
Most important, says Pignatti, is the need for trust and transparency to be at the heart of regulation, which is why the EMA is pressing ahead with a draft policy on publishing the clinical study reports on which it bases its assessments. “At present these are either not released or only with big redactions, and we feel we can address concerns about commercial confidentiality, inappropriate analysis and data protection. We think the release of these data can be managed so that it adequately addresses these concerns while allowing secondary analysis to scrutinise the regulatory process but, more importantly, to generate discovery about other factors such as prognostics, which will move forward the development of new drugs. Industry will have a lot to benefit from this as well.”
Tenhunen, who has commented on ‘how to assess assessments’ in Annals of Oncology (2013, 24:1138–40), says that regulators will have difficulties achieving “a delicate balance between transparency, legislation, interests of patients and healthcare professionals as well as those of the industry”. A ‘one size fits all’ regulatory approach will not work, he adds, especially with complicated new products such as Glybera (alipogene tiparvovec), the first gene therapy approved in the EU.
Tafuri’s prescription for better communication of processes and opinions includes the attendance of EMA regulators at FDA public hearings or of FDA staff at CHMP meetings. “This would certainly help mutual understanding of different regulatory systems and improve harmonisation.”
“With regard to transparency,” he adds, “all agencies should provide public access to the data and results from clinical trials on which regulatory decisions are based, and to committee minutes and public reports about the reasons why certain procedures for the approval of new active substances and indications result in either a successful or a failed application. Communicating the rationale of benefit–risk decisions to the public is crucial to promote trust in the regulatory system.”
PICTURE CAPTION (pg 15) On average the EMA takes around six months more than the FDA to approve a new drug or new indication for a drug. This is mainly due to time lost to clock stop and the delay between getting a positive CHMP opinion and approval from the European Commission. Furthermore, in the US almost all cancer drugs are approved under priority review, whereas accelerated assessment is rarely used by the EMA Source: CDER 21st Century Review Process (www.fda.gov); User Guide for Micro, Small and Medium-sized Enterprises (www.ema.europa.eu) *Day 150 for accelerated assessment; Rap – Rapporteur TABLE CAPTION (pg 17) Numbers indicate time to final approval from the European Commission, with time to positive opinion from EMA’s Committee for Medicinal Products for Human Use given in parentheses; b under review at time of publication; c Priority review procedure used Source: Adapted from RR Shah, SA Roberts and DR Shah. (2013) Br J Clin Pharmacol 76:369–411 Time to approval (days) for tkis in the EU and US


