



Traditional approaches to generating clinical evidence rely on recruiting large numbers of patients into trials. Paolo Bruzzi reflects on the challenges of designing and analysing clinical trials in rare cancers, and reviews the potential for using alternative trial designs and Bayesian statistical approaches to build robust evidence where patient numbers are small.
This grandround was first presented by Paolo Bruzzi, from the Institute for Cancer Research, Genoa, Italy, as a live webcast for the European School of Oncology. Paolo Casali, from the National Cancer Institute – IRCCS Foundation, Milan, Italy, posed questions raised during the e-grandround presentation. It was edited by Susan Mayor. The webcast of this and other e-sessions can be accessed at e-eso.net
The rarity of some cancers poses a challenge in conducting clinical trials with sufficient numbers to provide adequate power to assess the effects of a novel therapy. Rare cancers include: rare histologies in frequent sites, such as breast cancer with a squamous histology; cancers at rare sites, such as uveal melanoma; and cancers with both rare histologies and rare sites, such as astrocytomas and most sarcomas. Rarity is set to become a more general issue in cancer research, with growing interest in rare cancer conditions, increased recognition of rare presentations, such as skin metastases, and increased identification of molecular variants of many common tumours.

The ‘statistical mantra’ applied to clinical trials is that a study must have adequate size to provide adequate power to reduce the risk of false-positive or false-negative results, and to obtain precise estimates of the effects of the experimental therapy being investigated. The aim is to demonstrate a minimal difference that is considered clinically worthwhile, to a level of statistical significance (α) usually set at 5% (which means that out of 100 trials comparing treatments with identical effect on the primary endpoint, 5 will show a statistically significant difference by chance alone – that is they’ll provide a false-positive result). The power of a study (usually 80%–90%) indicates the probability it will obtain a statistically significant result, if the difference between the effects of the two therapies is the desired one.
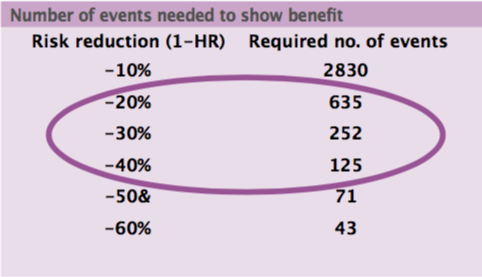
The minimal clinically worthwhile difference is usually a risk reduction, including mortality risk. The sample size needed in cancer trials for breakthrough drugs in early disease, based on cumulative mortality from 10% to 70%, is 500 to 5000 patients. In advanced disease, with cumulative mortality of 50% to 90%, the sample size required is 300 to 1000 patients. International co-operation is needed to gather a sufficient number of patients for a trial to have adequate size, but this may not be possible for some very rare cancers.
The assumption that a study must have an adequate size based on traditional statistical parameters can lead to the unjustified assumption that trials with small size are of poor quality.
Establishing therapeutic standards in very rare tumours/conditions
Where there are no trials, treatment of a rare tumour type may be based on ‘expert opinion’, although it is important to question what this is based on, or may be guided by indirect evidence. However, therapeutic standards can also be based on ‘small’ trials.
There are four key questions to consider when designing a small trial:
- Phase II or phase III?
- Randomised or uncontrolled?
- What are the endpoints?
- Conventional or unorthodox statistics?
Phase II or phase III trials?
If the number of patients is inadequate for a standard phase III trial, then it may be possible to run a phase II trial. There are several examples of phase II trials carried out in rare cancers over the past few years that have contributed important new information to their treatment and led to registration of new drugs based on comparing response rates against historical data. For example, a phase II, single-group trial of PD-1 blockade in advanced Merkel-cell carcinoma showed a median progression-free survival of nine months, compared to a historical value of three months (NEJM 2016, 374:2542–52).
Randomised or uncontrolled trials?
There are several false beliefs about randomised trials, including the myth that randomised trials require large numbers of patients, while uncontrolled trials do not. Second, some mistakenly think that uncontrolled trials do not require a statistical plan. The reality is that an uncontrolled trial, even one with appropriate statistical planning, is not necessarily the best option where patient numbers are smalll.
To conduct an efficacy trial in a rare condition, researchers must choose between internal validity – in which case a randomised trial is required – and feasibility – which means an uncontrolled trial (see figure above). Whichever type of trial is carried out, it is important to recognise and minimise sources of error. There are two types of error in any trial:
- Sampling error – due to chance. Preventing sampling error requires an increase in sample size.
- Bias – due to errors in selection of groups, assessment of outcomes or statistical analyses, which distort the evaluation of any associations observed. Methods to reduce bias include randomisation, masking (such as double blinding) and intention-to-treat analysis (all patients who were enrolled and randomly allocated to treatment are included in the analysis and are analysed in the groups to which they were randomised).
The benefits of an uncontrolled trial in a rare cancer are that it enables more patients to receive the new treatment being investigated, and it is easier to recruit patients.
A randomised trial provides unbiased estimates of treatment effects, but makes it more difficult to enrol patients, and fewer patients receive the new treatment being investigated.
It is important to remember that sampling error and bias are independent. Increasing sample size in the presence of bias can be misleading, because it gives researchers more confidence in a wrong result. Statistical methods deal mainly with sampling error but provide little help with bias.
If the expected, or necessary, treatment effect is large but not outstanding, then a randomised clinical trial, if ethically acceptable, is the best way to assess a new drug even in rare diseases. The advantages are validity and credibility, but the disadvantage is a moderate loss in power.
The problem is that there is often no standard treatment for a rare cancer, which may mean the control group is untreated, leading to issues around ethics and acceptability. As a consequence there may be situations where a randomised controlled trial may not be the best approach, or should be avoided for ethical reasons.
These include: when the prognosis with standard therapy is poor, or there is no therapy; when an experimental therapy is not very toxic; or when there is plausible efficacy for the experimental therapy, based on uncontrolled trials in the cancer being studied, randomised controlled trials in different stages of the same cancer, randomised trials in other cancers with the same biology, or dramatic effects having been observed in other cancers.
For example, the tyrosine kinase inhibitor imatinib was initially investigated in a large randomised controlled trial in chronic myeloid leukaemia (CML). The size of the effect was sufficient to suggest that randomised trials in rarer cancers were unethical, so the drug was evaluated in a much rarer cancer, gastrointestinal stromal tumour (GIST), with a large uncontrolled trial, and then with case series in other very rare indications, including dermatofibrosarcoma protuberans, plexiform neurofibromas, and chordomas.
New paths are emerging for investigating novel drugs (see figure opposite) that start with large randomised controlled trials in common cancers. These provide proof of principle and data on toxicity. The next step is uncontrolled but formal trials in other, often rare, cancers that share the same drug target. It is important to develop the best methodology for conducting uncontrolled trials, which should be rigorous and transparent and take account of biases, with much better selection and use of historical controls. Finally, a new drug can then be investigated in even rarer conditions with the same target with off-label use in individual cases.
Endpoints in cancer trials
There are two main types of endpoint in cancer trials: ‘true’ endpoints, including overall survival and validated quality-of-life scores, and surrogate endpoints, such as response rate and progression free survival.
None of these surrogate endpoints have been validated in rare cancers. However, objective response is reproducible and consistently associated with clinical benefit in solid tumours even without a control group, so is a preferred endpoint.
Progression free survival is sensitive to the type and timing of assessments and is meaningless without a control group, so always requires historical control data. Use of any surrogate endpoint in trials for rare cancers is acceptable only if the new treatment is associated with dramatic changes in prognosis, ideally in the long term.
Conventional or unorthodox statistics?
It is important to consider the expected frequency or probability of the event or observation being measured in a trial, given the hypothesis underpinning the trial. The statistical foundation of a randomised trial is based on the null hypothesis: that there is no difference between the two treatments being compared. Randomisation ensures that any differences between treatment groups are due to chance, and allocating treatment on a double-blind basis prevents bias in assessments. The trial tests whether the observed results are compatible with the null hypothesis that the two treatments being compared are identical.
With conventional (frequentist) statistics, the advancement of knowledge in medicine is based on assuming that the dominant theory is true (i.e. the standard treatment is better) until sufficient evidence becomes available against this. Only evidence collected within one or more trials aimed at falsifying the dominant therapy can be used.
The problem is that outstanding efficacy is seldom observed with new drugs in cancer, so this makes large trials necessary to show that the new treatment is more effective. In addition, this approach cannot make use of external evidence or evidence in favour of an alternative hypothesis. This means that any knowledge or results outside the primary analysis of a clinical trial is ignored in the trial’s design and analysis.

There are several recent developments in the design and statistical analysis of clinical trials to overcome these limitations, including: surrogate endpoints; new types of systematic review; and adaptive trials. Bayesian statistics provide one of the most important developments in trial analysis.
The concept of Bayesian probability is based on considering the probability that a hypothesis is true, given observation and prior knowledge. Frequentist probability looks at the probability of an observed difference based on the assumption that the experimental therapy being tested in a trial does not work. In contrast, Bayesian probability considers the probability that the experimental therapy works or does not work, given the observed difference and prior knowledge.
It is commonly thought that frequentist probability is objective and provides a ‘hard’ approach to analysing experiments, while Bayesian probability is subjective and ‘soft’. This is incorrect. They are simply different approaches to the meaning of probability and, most importantly, to the use of prior evidence, which is a key difference between conventional frequentist and Bayesian approaches. Frequentist probability makes no use of prior knowledge, whereas Bayesian probability makes considerable use of what is already known.
The disadvantages of Bayesian statistics are that they are considered to be somewhat subjective, arbitrary, and amenable to manipulation, with the fear that pharmaceutical companies could register drugs based on marginal benefits from trials. However, the conceptual advantages are that they reflect human reasoning, or ‘common sense’. The approach is focused on estimates of effect. It provides a conceptual framework for medical decision-making and, most importantly, is transparent because it makes explicit any assumptions made during interpretation of results.
There are practical advantages to using Bayesian statistics to analyse studies in rare tumours. There is no need to set the sample size in advance, facilitating adaptive trial designs in which patients are enrolled until there is sufficient evidence in favour or against efficacy. Where strong a priori evidence is available and trial results are in agreement with this evidence, then a smaller sample size is sufficient and a trial can be stopped earlier, when appropriate.
The critical factor for the use of Bayesian statistics is the availability of prior evidence, which should be transformed into a probability distribution. This evidence should be based on objective information using meta-analysis techniques drawing on a range of sources including randomised trials, biological and preclinical studies, case reports, uncontrolled studies, studies with surrogate endpoints, and studies in other similar cancers or in different stages of the same cancer.
Current approaches often make use of rational but informal integration of available knowledge. In contrast, use of Bayesian methods facilitates formal, explicit, and quantitative integration of available knowledge, using verifiable quantitative methods, sensitivity analyses and a focus on summary effect estimates.
In conclusion, a trial in a rare cancer requires more careful planning and protocol preparation than a trial for a more frequent cancer, because standard trial design and analysis techniques cannot be used. The design and methodology and the statistical analysis must be planned carefully in advance, making optimal use of available evidence.
Question: Can genetic subgroups be considered in a similar way to rarer cancers? Is precision medicine inevitably more imprecise, from a statistical point of view, because of small numbers?
Answer: The more you try to tailor a treatment to individual patients, the less evidence you have because the patient numbers are smaller. I think that Bayesian reasoning has always been used in clinical medicine, drawing on information from a range of settings, including nonrandomised trials, case series and past experience with patients. In molecular subsets of tumours we should make explicit use of Bayesian reasoning, using all information available from different settings. This is the core of Bayesian reasoning: what is the possibilty that this drug works in this group of patients?
Question: Regulators sometimes suggest setting up clinical registries of patients with rare cancers as a source of external controls, which we often lack when we plan uncontrolled studies. Are there methodologies for doing this?
Answer: I think patients referred to specialist centres should have their data included in registries. There is a good example of this kind of registry in Italy, but it has not yet provided cases that are useful for proving efficacy. What we need to do is select data from unselected registries for cases that are comparable to those included in trials, and we need unselected historical controls. It is important to remember that, when we introduce a new treatment, the prognosis of subgroups changes. For example, cases of sarcoma today do not have the same prognosis as patients seen 10 years ago. The best approach is to maintain a population registry and to be able to abstract cases eligible for a new drug from that population base. Artificial intelligence could be used to interrogate large databases to identify historical controls.
Further reading
A consensus position on a set of methodological recommendations for clinical studies in rare cancers, developed by Rare Cancers Europe, co-authored by Paolo Bruzzi, was published in the Annals of Oncology (2015, 26:300–6).


