Pharmacokinetics within tumour cells play an important role in the development of resistance. A better understanding of the mechanisms involved is important for devising treatment strategies to extend tumour response.
 We know that cancer cells develop acquired capabilities, including an ability to evade apoptosis, and develop insensitivity to anti-growth signals (Cell 2000, 100:57–70). We have less information, however, on how cancer cells are able to develop resistance to drug action by bypassing drug signalling and also decreasing drug levels at the target site.
We know that cancer cells develop acquired capabilities, including an ability to evade apoptosis, and develop insensitivity to anti-growth signals (Cell 2000, 100:57–70). We have less information, however, on how cancer cells are able to develop resistance to drug action by bypassing drug signalling and also decreasing drug levels at the target site.
Adsorption, distribution, metabolism and elimination (ADME) are the key processes underlying the pharmacokinetics (PK) of any drug, each of which may be changed during the development of resistance. The figure overleaf illustrates intratumoural ADME in the development of drug resistance, summarising how drugs can react in cells during cancer treatment. The starting point is a tumour, comprising a population of different clones, which is treated with a drug. Hopefully, a lot of the cancer cells go into apoptosis and die. However, there are often resistant clones that rapidly lead to primary resistance. In a second scenario there is selection of tolerant clones that will show growth stabilisation. These cells are apparently dormant, but in reality they are activating resistance mechanisms designed to help the cells overcome the action of the drug. After a few months or years, these dormant cells develop secondary resistance to the drug, so it is no longer effective.
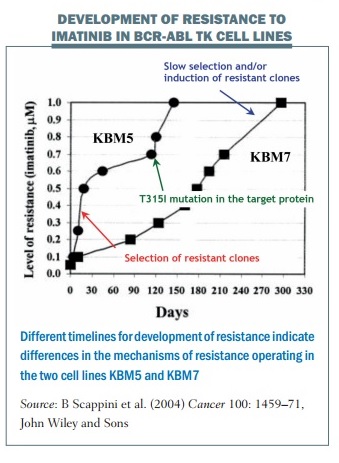
The figure below shows two cell lines that illustrate different mechanisms of development of resistance to imatinib over one year. Resistance develops over different time periods in the two lines. The first cell line (KBM5) shows rapid development of resistance by selection of pre-existing resistant clones. After about 120 days, new clones appear with a mutation in the target protein for imatinib (Bcr-Abl), which confers resistance. In contrast, resistance develops in a stepwise manner in the second cell line (KBM7), without a strong, clear event. This probably indicates the slow induction of different resistance mechanisms that are selected for, so in the end the resistance is as high as in the first clone.
The six golden rules in intratumoural ADME
 Cancer cells fight back against drug treatment because they are armed to survive. In a review I wrote several years ago (Curr Cancer Drug Targets 2009, 9:652–674), I proposed ‘six golden rules’ in intratumoural ADME:
Cancer cells fight back against drug treatment because they are armed to survive. In a review I wrote several years ago (Curr Cancer Drug Targets 2009, 9:652–674), I proposed ‘six golden rules’ in intratumoural ADME:
Rule 1: Pharmacokinetics in the blood are different from the intratumoural pharmacokinetics.
An example would be two patients with the same plasma drug levels but very different drug levels in the tumour, so the patient with the lower intratumoural drug level will have a higher risk of cancer relapse.
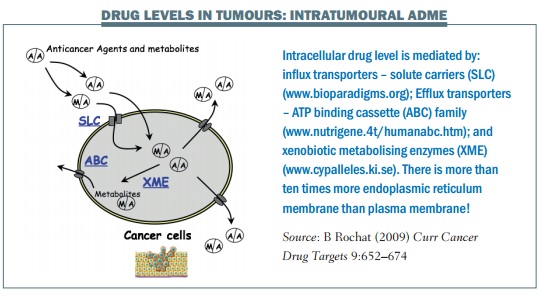
Rule 2: There are three main systems involved in drug disposition in cancer cells (see figure below):
- influx of the drug into the cell (SLC channel)
- efflux of the drug out of the cell (ABC pumps)
- degradation of the drug by xenobiotic metabolising enzymes (XME).
A drug targeted to a cancer cell may enter through a channel in the membrane, using solute carriers (SLC). Efflux transporters such as the ATP binding cassette (ABC family) may take the drug or its metabolites out of the cell. The drug may be degraded inside the cell by xenobiotic metabolising enzymes, typically cytochrome P450 enzymes (CYP). There is over ten times more endoplasmic reticulum (ER) membrane than cell membrane in tumour cells, providing a large volume for CYP metabolising enzymes.
 Rule 3: There is synergistic interplay between these three systems
Rule 3: There is synergistic interplay between these three systems
SLC, ABC and XME – that has been built over hundreds of millions of years of evolution. A drug can enter the cell, bind to the nuclear receptor and direct the DNA to increase the number of efflux pumps as well as CYP or other xenobiotic metabolising enzymes. The same receptor can increase both efflux and degradation enzymes inside a cell.
In a sensitive cancer cell, the drug enters the cancer cell and carries out its action that kills the cell. However, eventually the cell fights back by reducing influx and increasing efflux and CYP enzymes, so there is a much lower level of the drug (e.g. ten times less drug) inside the cell.
Rule 4: There is great variability in the expression of these three systems between tumours.
This occurs because DNA is highly unstable in cancer cells, with high rates of mutations and activation of transposons (mobile DNA sequences). Under the stress that an anti-cancer drug can impose on a cell, the transposons can activate different CYP enzymes to be overexpressed inside the cell (see, for instance, Cancer Res 2005, 65:3726–34). The figure below shows glucuronidation activity in normal colon biopsies (white bars) compared to that in cancer biopsies (grey bars), illustrating the difference in activity in different patients. The much higher enzyme activity in some patients explains why their intratumoural drug levels will be much lower than in those with lower glucuronidation activity. Similar variability – up to a ten-fold difference or more – is seen in influx and efflux proteins.
 Rule 5: Intratumoural CYP can play a role in anticancer drug degradation and the synthesis of messengers involved in cell survival or proliferation.
Rule 5: Intratumoural CYP can play a role in anticancer drug degradation and the synthesis of messengers involved in cell survival or proliferation.
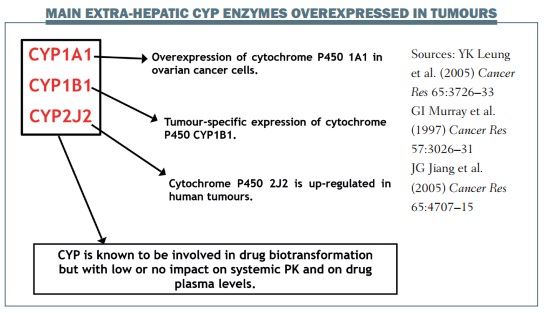
Certain CYP enzymes, such as CYP1A1, 1B1 and 2J2, are poorly expressed in the liver or intestine, but can be overexpressed in many tumours. CYP enzymes expressed in the liver are the ‘canonical’ enzymes and are studied extensively, especially by pharmaceutical companies. Those expressed in cancer cells are the ‘exotic’ enzymes, and are poorly studied. Many extrahepatic CYP enzymes are overexpressed in tumours, with the potential to affect intratumoural pharmacokinetics only.
Rule 6: The three systems – SLC, ABC and XME – are all involved in the appearance of drug resistance.
This was shown, for example, in a study of mice with two types of tumour – wild type with normal efflux activity, or null ABCG2 (BCRP) genotype with lowered efflux for topotecan. Resistance to the topotecan developed much faster in the wild type mice than in those with deletion of the ABCG2 transporter. The first conclusion was that each tumour was unique in its response to the therapy; the second conclusion was that efflux was involved in resistance, but this was a transient event (PNAS 2007, 104:12117–22; S Rottenberg, Biomedical Transporters conference 2007, Bern, Switzerland).
Why study intratumoural pharmacokinetics?
Pharmaceutical companies study the pharmacokinetics of cancer drugs, but focus on only one aspect – the metabolites produced by the liver and not by the cancer cells. We recently discovered more than 40 metabolites of tamoxifen circulating in the plasma of treated patients (Anal Bioanal Chem 2014, 406:2627–40). This example shows that it is important to remember that metabolising enzymes have strong efficacy in degrading drugs, but that their intratumoural role is generally ignored.
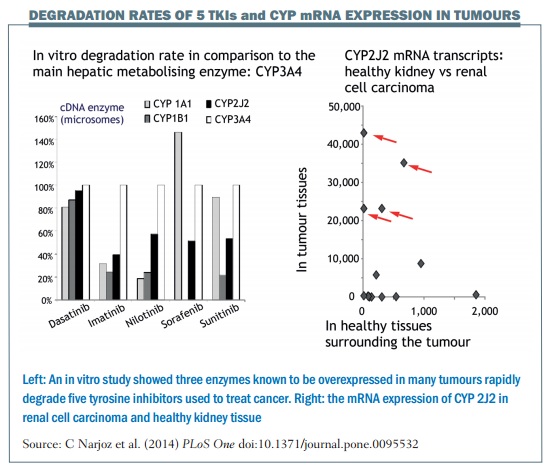
We recently studied the role of three extra-hepatic P450 enzymes – CYP1A1, 1B1 and 2J2 – which are known to be overexpressed in many tumours – in the degradation of dasatinib, imatinib, nilotinib, sunitinib and sorafenib. Results showed that these three extra-hepatic CYP enzymes had strong affinity (Km) and degradation velocity (Vmax) for the five tyrosine kinase inhibitors (TKIs) tested. Degradation efficiencies were comparable to the major hepatic CYP, CYP3A4 (see figure below). We looked at the RNA expression of the enzymes in patients with renal cell carcinoma and hepatocellular carcinoma, in tumour biopsies as well as in their healthy tissue counterpart.
 Results showed that CYP2J2 RNA was overexpressed in about one-third of the tumours, suggesting a probable high degradation of TKIs by CYP2J2 in these tumours.
Results showed that CYP2J2 RNA was overexpressed in about one-third of the tumours, suggesting a probable high degradation of TKIs by CYP2J2 in these tumours.
What are the clinical consequences of intratumoural drug metabolism?
CYP2J2 is highly expressed in haematological cancers and promotes tumour cell growth, proliferation and metastasis. It is up-regulated in many human tumours, including breast, stomach, oesophagus, liver, colon and lung (J Pharmacol Exp Ther 2011, 336:344–355; Cancer Res 2005, 65:4707–15, Cancer 2009, 28:93–96; Life Sci 2008, 83:339–345; Cancer Res 2007, 67:6665–74). CYP2J2 has been demonstrated to rapidly biotransform various TKIs (PLoS One 2014, doi:10.1371/journal. pone.0095532).
All of this suggests that CYP2J2 could be a good target enzyme to inhibit because it probably activates promoters of cancer cell growth as well as degrading TKIs. From a clinical perspective, there are already a few approved drugs known to be strong inhibitors of CYP2J2, including telmisartan, flunarizine, danazole and amiodarone. Using these drugs to inhibit CYP2J2 could provide a novel strategy to improve TKI efficacy and extend the time to relapse. It is similar to inhibiting beta-lactamase metabolising enzymes to increase drug exposure and reduce antibiotic resistance. I think this offers a promising approach for reducing the development of cancer drug resistance.
 Daniel Helbling, of the Gastrointestinal Tumour Center in Zurich, Switzerland, hosted a live question and answer session
Daniel Helbling, of the Gastrointestinal Tumour Center in Zurich, Switzerland, hosted a live question and answer session
Q: Do you think the pharmacokinetic mechanisms of resistance that you covered are the mainstays of all resistance?
A: No, there are many different mechanisms by which cancer cells can develop drug resistance. But this is one mechanism we should think about, and consider inhibiting to increase intratumoural drug levels. It is similar to what happened 10 years ago when people worked on P-gp inhibition (ABCB1 efflux pump), with clinical trials using inhibitors of ABC transporters. Unfortunately, this turned out not to be possible because these inhibitors also affected healthy cells (e.g. in the liver or at the blood–brain barrier). This is different for CYP2J2, because it is expressed mainly in cancer cells, and only very poorly in healthy liver cells.
Q: Are there any case reports of adding CYP2J2 inhibitors to cancer treatments showing increased efficacy?
A: No. I think people have considered giving 2J2 inhibitors not as inhibitors of drug degradation but rather as inhibitors of messengers that are promoters of cell proliferation and metastasis. However, both mechanisms could potentially be targeted with the same inhibitors. This is ongoing, I believe.
Q: Is there any way to predict intratumoural pharmacokinetics?
A: It is a good question, and we should be able to do this. I tried to contact people in Geneva where they have computer models using kinetic parameters such as affinity of the enzymes for the drug and different compartments to simulate the pharmacokinetics in the whole body, in the plasma and in the tumour. The software is not designed for modelling pharmacokinetics in tumours, but I think it should be possible, and the group wants to try it.
Q: Does radiotherapy influence intratumoural pharmacokinetics?
A: As far as I know radiotherapy kills cells so there will be no pharmacokinetics or degradation capability in dead cells. The other question would be whether radiotherapy activates transposons and maybe modifies DNA stability. I do not think that radiotherapy would be a feasible approach to modifying intratumoural pharmacokinetics. It is difficult to look at what is happening in the tumour, especially in a human, although it may be possible with an animal model. It has been looked at in the opposite way, using gene therapy for the CYP involved in activation of the pro-drug in the tumour (e.g. the alkylating agent cyclophosphamide), with the aim of using a lower dose of drug to reduce side-effects or increase intratumoural drug levels. This has been shown to be effective and promising in animal models.
Q: Do we know the drugs that are affected or prone to being metabolised by CYP2J2 enzymes?
A: There are only a few drugs that are considered to be substrates of CYP2J2. Why? Because in the liver the level of this enzyme is very low, and other CYP enzymes are involved in drug metabolism. There are a couple of drugs that are not biotransformed by usual hepatic CYP enzymes, including 3A4 and 1A2, but are biotransformed by CYP2J2 (weakly expressed in the intestine). In vitro experiments show that CYP2J2 is able to degrade a lot of drugs, but, of course, this is not in the liver. However, where it is expressed, such as in many tumour tissues, it can be a strong enzyme in drug degradation.
Q: Which anticancer drugs that we use every day would be most prone to have a better efficiency by giving a CYP2J2 inhibitor?
A: According to our results, with almost all of the TKIs we tested, CYP2J2 is as efficient, or very close to being as efficient, as 3A4, which is the most important enzyme in the liver in terms of degradation of drugs. Expression of 2J2 depends on the tumour. In a prospective study, we looked at CYP2J2 overexpression in 14 tumour biopsies and their healthy counterparts. One-third showed a very high CYP2J2 expression, which was compatible with high intratumoural drug degradation. If the enzyme is present then you could expect that most TKIs would be rapidly degraded, specifically in the tumour.
Q: Would this pave the way for clinical studies?
A: I think and hope so. Some caution would be needed because CYP2J2 is expressed in the heart, so you would not want patients to be to highly exposed to an inhibitor for an enzyme that plays a role in the heart. However, several drugs that are inhibitors of this enzyme are already on the market (telmisartan, flunarizine, danazol and amiodarone), and are used in patients for chronic treatment, with an acceptable safety profile.