Recent years have seen a revival of interest in the unique metabolism of cancer cells. This time the focus is on the potential it offers as a target, rather than any possible causal role but that link with obesity still needs explaining.
 The association between cancer and altered cell metabolism was first highlighted by a German biochemist more than 90 years ago. Otto Warburg observed in 1924 that cancer cells process glucose – one of the body’s key nutrients – into lactate, as athletes’ muscles do when they run short of oxygen, but they do so even when they have sufficient oxygen. They also process glucose much faster than normal cells, which rely mostly on using mitochondria as the engine room for producing energy.
The association between cancer and altered cell metabolism was first highlighted by a German biochemist more than 90 years ago. Otto Warburg observed in 1924 that cancer cells process glucose – one of the body’s key nutrients – into lactate, as athletes’ muscles do when they run short of oxygen, but they do so even when they have sufficient oxygen. They also process glucose much faster than normal cells, which rely mostly on using mitochondria as the engine room for producing energy.
The Warburg effect, as it became known, was put forward by its proposer as the cause of cancer, resulting from the impairment of mitochondria. The effect has been widely discussed and the biology explored, but after a while the field of cancer research moved on.
Interest in the metabolism of cancer cells revived with the advent of PET scanning using radiolabelled-glucose, in the early 1980s. This time, however, the focus was on making use of the Warburg effect to obtain images of the behaviour and spread of an individual cancer.
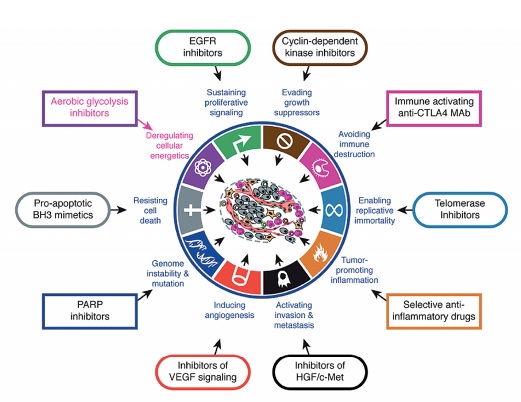
Source: D Hanahan and RA Weinberg (2011) Hallmarks of cancer: the next generation. Cell 144:646–674, reprinted with permission from Elsevier
But despite the known metabolic actions of some of the most widely used chemotherapy drugs, such as methotrexate and 5-fluorouracil (5FU), there was little interest in addressing metabolism as a possible weak link in cancer that could be targeted. The famous ‘hallmarks of cancer’ paper by Douglas Hanahan and Robert Weinberg, from the year 2000, for instance, did not include metabolism.
That’s all changed now, and the Warburg effect is again centre stage. And although most researchers do not believe it causes cancer, there is great interest in whether this and other metabolic changes in cancer cells might be an Achilles heel that could be targeted. Now, new knowledge about the function of cancer genes and their relationship with the way cells metabolise a range of nutrients, together with epidemiological evidence from the relationship between obesity, exercise and cancer, is fuelling a rapidly growing cancer metabolism field.
This has reached the point of dedicated international research conferences, a new journal, a growing number of investigational agents in both public and private research, and some clinical trials, in particular of the low-cost antidiabetic drug, metformin. And in 2011 Hanahan and Weinberg updated their ‘hallmarks’ to include abnormal metabolic pathways.
Oncogene–metabolism cross-talk
“Back in the early 1990s it was assumed we knew all about metabolic pathways of glucose and amino acids – they were set out in standard biochemistry textbooks,” says Chi Van Dang, director of the Abramson Cancer Center, University of Pennsylvania, and a medical oncologist who researches cancer metabolism.
“We thought that it was only cancer genes that drive the cell cycle machinery that cause cells to replicate, and the energy part was just along for the ride and doesn’t need to be regulated. But what we have found is that there are specific oncogenes that drive many other genes, including metabolic ones, rather like an orchestra conductor, both causing cells to replicate and to take in nutrients to do so.”
This was initially met with scepticism, he says, but it is now known that, somehow, cancer genes ‘cross-talk’ with metabolic pathways. It is not the case that when cancer genes send the signals that turn on the DNA replicating machinery, the energy to carry out that proliferation is there as if by magic. “Many cancer genes cross-talk with machinery that brings in glucose or amino acids,” says Dang, who says a similar shift in knowledge took place with angiogenesis, where it is now known that a tumour can release hormones to grow new blood vessels to feed itself.
“Many cancer genes cross-talk with machinery that brings in glucose or amino acids”
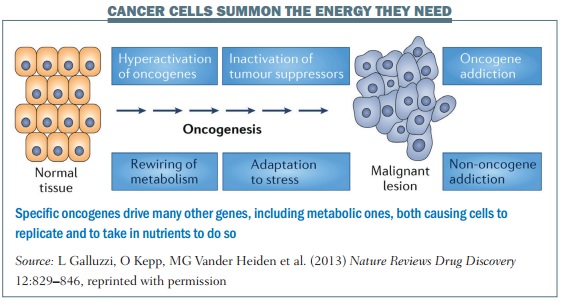 Dang likens the behaviour of normal cells to the way a tall building is constructed – materials such as bricks and cement have to be shipped in an orderly fashion and coordinated, or orchestrated, for growth. A normal cell has feedback loops that tell it that, if it doesn’t have enough nutrients or oxygen, it won’t divide, as it could make genetic mistakes. Only when conditions are right will it build up to divide in an orderly way with the least chance of a genomic error.
Dang likens the behaviour of normal cells to the way a tall building is constructed – materials such as bricks and cement have to be shipped in an orderly fashion and coordinated, or orchestrated, for growth. A normal cell has feedback loops that tell it that, if it doesn’t have enough nutrients or oxygen, it won’t divide, as it could make genetic mistakes. Only when conditions are right will it build up to divide in an orderly way with the least chance of a genomic error.
“In a cancer cell the same switches, instead of turning on and off, are permanently on, owing to genetic mutations, such as with the Myc oncogene that we study in my lab, and you have a deregulated system that continues to grow without the right nutrients. But that also creates a vulnerability, because the cells are addicted to nutrients. It’s like building a wall with bricks but without cement.”
Epidemiological evidence
If the data from cell biology are becoming compelling, relating the knowledge to observations about animals, humans and cancer rates adds important context about the causes of cancer. When cells divide and there are mistakes or mutations in the process, the accumulation of these mutations can lead to cancer if they permanently turn on a cancer gene.
But as Dang points out, there is a paradox about cancer in animals, named after British epidemiologist Richard Peto. Given that large animals have many cell divisions to grow to adulthood – and elephants certainly grow to a large size – it would be expected that they would also suffer from a higher cancer rate than smaller animals, such as the much-studied mouse.
“But elephants have much lower cancer rates than mice and the answer lies in the metabolic rate of their cells – mice have a rate a hundred times greater than elephants, which of course also live much longer,” notes Dang. If mice are given drugs to restrict metabolism, such as metformin, which slows mitochondrial function, or rapamycin, which inhibits a growth and metabolism pathway, they live longer. “And a recent study shows that if you remove just one copy of the Myc gene in mice, metabolism is slowed and that can prolong their life. The interpretation is that a higher metabolic rate increases mutation rates and contributes to cancer development.”
“The interpretation is that a higher metabolic rate increases mutation rates and contributes to cancer development”
In turn this leads to the idea of ‘metabolic fitness’ and relationships between cancer, obesity, diet and exercise. The evidence for a link between obesity and cancer is now a given. The US National Cancer Institute, for example, projects that, by 2030, there will be 500,000 additional cancer cases in the US owing to obesity, with the risk for some cancers, such as oesophageal, pancreatic, colorectal and endometrial, increasing more than for others.
Where’s the link?
There is though no single explanation of the causal mechanisms linking people with high body mass index (BMI) and cancers – so far, molecular mechanisms are poorly understood. Dang says research is ongoing into calorific restriction, which can increase metabolic fitness – fewer nutrients can lower the metabolic rate – and factors such as hormones that could promote cancer.
One explanation for the cancer–obesity link is that higher glucose levels in overweight people are caused by insulin resistance, or a form of type 2 diabetes. “Cells cannot respond to glucose as well, so the body simply makes more insulin and an insulin-like growth factor (IGF-1), which are believed to drive cancer cells to a more aggressive state. The thinking is that it’s not glucose on its own, and this model fits with a lot of data we have,” he says.
A recent study adds evidence about the role of insulin. It found that postmenopausal women who are overweight but ‘metabolically healthy’ are not at elevated risk of breast cancer compared with women who are metabolically healthy but have a normal weight. However, women with high insulin levels have a higher breast cancer risk whether they are normal weight or overweight (and being overweight is in turn a risk factor for insulin resistance). The researchers suggest that “metabolic health may be more biologically relevant and more useful for breast cancer risk stratification than adiposity per se” (J Cancer Res 2015, 75:270).
“Metabolic health may be more biologically relevant… than adiposity per se” Other possible mechanisms include higher oestrogen production by fat tissue – hormones are of course implicated in several cancers – or inflammation, which can also have metabolic components. A recent special issue of BioMed Research International highlights a growing awareness of the link between altered cellular metabolism and the risk of developing diabetes and cancer. It includes papers that show that key pathways of fatty acid metabolism are altered in cancer, and that there is a strong prevalence of cancer in postmenopausal obese women, with associations between obesity, ovarian steroid hormones and cancer. The same issue also looks at the promise that some drugs used to treat diabetes, such as metformin, are showing as cancer therapies, and the possibility of targeting insulin growth factors.
The biochemical analysis in these papers is complex, but as Dang says, the field is simplified somewhat by the fact that cells use only a small number of major nutrients – principally glucose, glutamine and fatty acids/lipids, although other substances such as acetate are also important.
One probable advantage of targeting metabolism may therefore be that approaches may extend across a range of tumours, owing to common biochemistry, although Dang points out that not all cancers are addicted to the same nutrients: “Breast cancer is addicted more to glucose, for example, and pancreatic cancer to glutamine.”
Treatment opportunities
The search for therapeutic drugs is focusing particularly on the metabolic pathways by which nutrients are used by cancer cells, and enzymes in these pathways that could be inhibited. Enzymes are targeted in plenty of other drug applications, but in the cancer metabolism field, research into questions such as how enzymes operate in the glucose pathway is in its early stages.
Metformin, the drug used in diabetes to control blood sugar levels, is one of the most investigated so far in cancer metabolism. Although the exact mechanisms are still being researched, says Dang, it is known to act on an enzyme target called complex I, the first enzyme in the mitochondrial energy chain used to generate ATP (adenosine triphosphate, which every biology student knows as the key energy transfer chemical), thus slowing down the ability of cancer cells to breathe so they can’t burn up nutrients.
This works because Otto Warburg’s original hypothesis, that cancer cells are glycolytic and don’t use mitochondria much, has been superseded by research showing that most cancer cells do, in fact, need mitochondria and do breathe oxygen. Metformin can also work against cancers by insulin control.
As metformin has been prescribed to millions of people for many years, and is known to be very safe, there are fewer obstacles to using it in clinical trials, usually in combination with chemo- or targeted therapies. “We are seeing a real biological effect from metformin in clinical trials,” says Dang, and there are already data suggesting that people with diabetes who take metformin have a lower risk of developing cancer or dying from it.
Statins are another group of cheap and widely used drugs that are attracting interest for potential use against cancer, as tumours are known to need to synthesise their own cholesterol. There are now a number of trials of statins in prostate cancer, as well as retrospective analyses comparing rates of prostate cancer incidence and survival between men who have been on statins, and those who have not.
Therapeutic targets and drugs in trials
- A number of drugs aimed at metabolic targets are now in early trials for a variety of cancers, including: AZD3965, a monocarboxylate transporter (MCT1) inhibitor being trialled in patients with advanced solid tumours
- DCA (dichloroacetate), a PDK1 inhibitor, being trialled in patients with recurrent malignant brain tumours, metastatic breast cancer and advanced non-small-cell lung cancer
- TCD-717, a choline kinase inhibitor being trialled in patients with advanced solid tumours
- AG-221, an isocitrate dehydrogenase (IDH) inhibitor being trialled in certain advanced solid tumours, including glioma, and with acute myeloid leukaemia and angioimmunoblastic T-cell lymphoma (AITL) with IDH mutations
- The diabetes drug metformin, being trialled for use in a number of cancer settings, including as a preventive in overweight or obese premenopausal women with metabolic disturbances, as an adjuvant in patients treated for early breast cancer, and in patients with advanced refractory colorectal cancer
- Statins, used to lower cholesterol, being trialled for use in a variety of cancers including prostate, colorectal and breast cancer, in therapeutic, preventive and adjuvant settings.

A therapeutic window
The key, as always, is to find ways of attacking cancer cells that don’t harm normal cells, says Almut Schulze, a professor at the department of biochemistry and molecular biology, University of Würzburg, Germany, and co-chair of an American Association of Cancer Research (AACR) meeting on cancer and metabolism.
With metabolic approaches, one aim is to find interventions that inhibit cells’ metabolic activity and their need to proliferate such that they die, while normal cells may slow down to a resting state, and are much less susceptible to this inhibition.
One aim is to find interventions that inhibit cells’ metabolic activity such that they die
“The difference with targeting metabolism and using targeted therapies such as imatinib [Glivec] is that we are not attacking proteins or genes that are genetically changed in cancer cells, but other factors needed for proliferation. It’s what we call non-oncogene addiction.”
She adds that a big problem in targeting oncogenes, such as BRAF in advanced melanoma, is the rapid development of resistance. However, when people relapse there is also a change in metabolism in cells that could be addressed with new metabolic inhibitors, used in combination with existing therapies, which could be drugs or radiotherapy.
There is also a need to distinguish between the metabolism of normal proliferating cells, such as hair follicles, skin and the gut lining, and cancer cells (chemotherapy affects these normal cells as well).
Schulze’s own research group, for example, is focusing on lipids, molecules that include fats and are used as building blocks in cell membranes. Normal tissues receive lipids from the blood after synthesis in the liver, she says. “But we know that tumours start synthesising lipids from sugar – what advantage is that and why don’t they use blood lipids – and can we inhibit this? As most tissues don’t synthesise lipids we could intervene in tumour growth without affecting other tissues too much, although the liver may be at risk from toxicity with such an agent.”
Dang and colleagues, meanwhile, have demonstrated the fundamental role of the Myc oncogene – in a seemingly simple experiment, they put Myc in normal cells and then compared what happened when glucose was removed from their nutrients by doing the same with cells without the gene. “Normal cells go to rest, but the Myc cells die because you take away a building block and they crash from metabolic death – they try and keep up with the energy demand that their machinery needs.”
“Normal cells go to rest, but the Myc cells die because you take away a building block” There are many other targets in the various pathways under investigation, and in two main types of metabolism: bioenergetic metabolism, such as with metformin and mitochondria, and anabolic metabolism, which is about building cells, as with lipid synthesis. Hypoxia, the lack of oxygen commonly seen in tumours, is also a big factor in the metabolic picture, and it also drives angiogenesis – the promotion of blood vessels to bring in oxygen and nutrients – so there is now strong interest in the interplay between these functions.
Other enzymes under investigation include one Schulze has been involved with, which uses acetate in metabolic processes and has been found to be essential for cancer cells. “If we can disrupt it the cells can’t grow,” she says, adding that the work is a collaboration with AstraZeneca, while a competing study has already moved to using an investigational compound.
Dang mentions that a major advance has been made in cancers that have mutations in certain enzymes, where a drug can turn off the abnormal enzyme. There are early trials using this approach in acute myeloid leukaemia as well as preclinical data for certain brain tumours, using isocitrate dehydrogenase (IDH) metabolic enzymes.
Like most other cancer fields that are on the verge of new therapies, a lot of the activity is in the US, and specifically in the Boston area. One company that is betting on cancer metabolism is Agios, which has two IDH inhibitors at phase I and another agent entering phase II. Activity is more fragmented in Europe, but a particularly strong academic centre is the Beatson in Glasgow (where, in 1896, George Beatson made the first observation of the link between hormones and breast cancer).
It is still early days for cancer metabolism, and Dang says the data are likely to prove some parts of the thinking right but some wrong. “But it’s exciting as we can probably create a whole new class of drugs – although there won’t be a silver bullet as they are unlikely to work on their own.” Says Schulze: “There is a lot of promise but we really do need some results from the first drugs now. The initial hype is over.”
“There is a lot of promise but we really do need some results from the first drugs now. The initial hype is over”
Apart from new therapies, new knowledge about metabolism also has implications for public health messaging about diet, obesity and exercise, such as with the latest dietary guidelines in the US, which have relaxed on cholesterol intake but are more strict on saturated fats and sugar.
There is low public awareness of the link between obesity and cancer, and some researchers are urging new multidisciplinary work to tackle the problem. A new term – ‘adiponcosis’ – has been proposed for the condition by Italian researchers (J Clin Endocrin Metab 2013, 98: 4664–65).
And emerging from this highly complex picture is a particularly controversial point from some researchers – that the paradigm of cancer as a genetic disease is wrong and that it is actually primarily a metabolic disease, with all recognised cancer hallmarks being ‘downstream’ from the “initial disturbance of cellular energy metabolism” (see Seyfried et al. Carcinogenesis 2013, 35:515–527). They are at least asking the age old chicken and egg question: Which comes first, cancer cells or abnormal metabolism?
Building the Metabolic picture
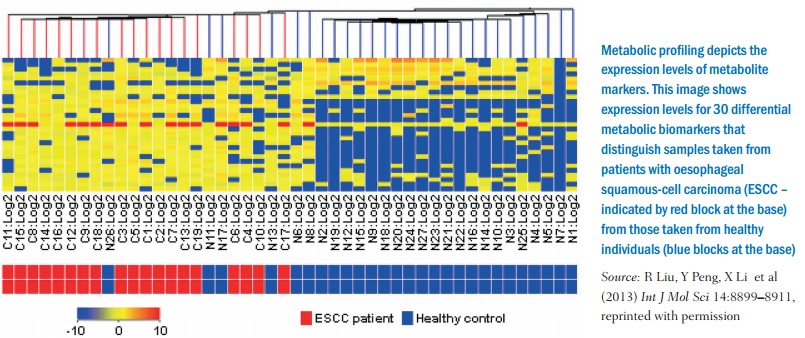
Much of the research into targeting the metabolic processes of cancer cells is only now possible because of techniques such as screening genes to reveal more metabolic functions, metabolomic profiling, which can identify metabolites from abnormal pathways in cancer, and also the rise of systems biology to model metabolic processes, as they can be interconnected in a widespread network and looking at one process in isolation could be insufficient.
One important area for research is so-called whole-body metabolism, as there are limitations to using laboratory cell cultures and animal models – and that is an obvious way forward given that the very first application of the Warburg effect uses PET to highlight metabolism in cells in the body (in vivo), and there are now other functional imaging methods and ways to measure metabolites in people. A new paper by Jared Mayers and Matthew Vander Heiden, ‘Famine versus feast: understanding the metabolism of tumors in vivo’ sets out the stall: “Examining tumor metabolism in vivo introduces new complexities, but taking this step is crucial to gain a deeper understanding of how whole-animal physiology impacts nutrient availability, as well as to appreciate the role of tumor heterogeneity and interactions between different cell types in tissues.” They make observations about how, for example, “pancreatic cancer can alter whole-body metabolism, causing new onset diabetes and cachexia in many patients”; there is “metabolic cooperation between different populations of cells within tumors” and “metabolic interactions with non-malignant tumor stromal cells can also directly influence disease progression, metastasis and redox [reduction-oxidation] status.” (See Trends Biochem Anal 2015, 40:130–140.)
There are many papers now on cancer and metabolism, notably review style write-ups such as ‘Famine versus feast’, which although highly technical may also have glossaries and break-out material, a clear indication that this field is in briefing mode about current thinking (see also ‘Metabolic targets for cancer therapy’, Nature Rev Drug Discovery 2013, 12: 829–846 for another good paper). There are also plenty of recent papers that revisit and explain the Warburg effect (e.g. Mol Biol Rep 2015, 42:819–823).
Dang and colleagues launched a journal, Cancer & Metabolism, in 2013, and there are now several research conferences, such as Metabolism and Cancer, run by the American Association for Cancer Research (AACR), which will be held on 7–10 June this year in Washington DC.