Clifford Hudis takes a look at how dose-dense regimens, intended to increase efficacy by decreasing the time between doses, work and effect toxicity in early breast cancer, especially where targeted agents are added.
Dose-dense regimens are intended to increase efficacy, not by increasing the patient’s total exposure to a drug, but by decreasing the time between doses. Does it work? And what happens to toxicity, especially where targeted agents are added? Clifford Hudis takes a look at the evidence in early breast cancer.
Why escalate the dose of cancer therapies? The rationale is that escalating the dose should kill more cancer cells. This has been seen many times in preclinical models in laboratory experiments and sometimes in the clinic, but not consistently. For example, two large, randomised trials, including a total of nearly five thousand patients in the NSABP (National Surgical Adjuvant Breast and Bowel Project) in the US showed no effect of escalated doses of cyclophosphamide on outcomes. These trials tested five dose levels, where the dose was doubled in dose size, doubled in dose exposure, doubled in dose size again, and doubled in total exposure again, so that doses ranged from 600 mg/m2 every three weeks to four times greater (see figure below). Results showed no impact on either disease-free or overall survival across these two sequential studies.
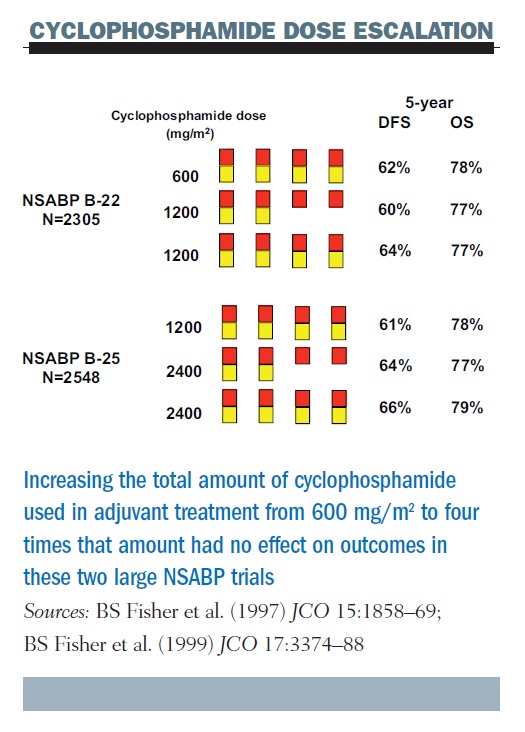 Previous results, such as those from Budman et al. (CALGB 8541) suggest that there could be a dose–response relationship for cyclophosphamide, but only at lower doses; the NSABP data show that this does not continue at higher doses. We have seen similar results for anthracyclines and taxanes and most other chemotherapy drugs.
Previous results, such as those from Budman et al. (CALGB 8541) suggest that there could be a dose–response relationship for cyclophosphamide, but only at lower doses; the NSABP data show that this does not continue at higher doses. We have seen similar results for anthracyclines and taxanes and most other chemotherapy drugs.
In optimising chemotherapy regimens with regard to dose and schedule, there are essentially two aspects to consider. On the one hand there is the Gompertzian growth kinetics of breast cancer cells, as is true for all other solid tumours, and indeed all cell and tissue types. The tumour, while always growing, appears to have a decreasing rate of growth over time. This is not actually true when you look at raw numbers, but it is true when you look at volumes. That is because of the effect of three dimensions in minimising the perception of volume change. It is also a reflection of the balance (or imbalance) between cell division and cell death as it changes with tumour growth, perhaps due to alterations in the delivery of nutrients and other factors.
If we administer chemotherapy based on the Skipper–Schabel model (see figure, below), the green arrows indicate the further reduction with each dose of chemotherapy, which we have always been taught is a log kill effect. The black arrows show the result of shortening the time between treatments on the log kill effect, which is what we call dose density. More frequent (dense) dosing decreases the time for tumour regrowth in between doses. It allows for the treatment each successive time of an ever smaller volume of tumour and that, in turn, results in a greater overall cell-kill.
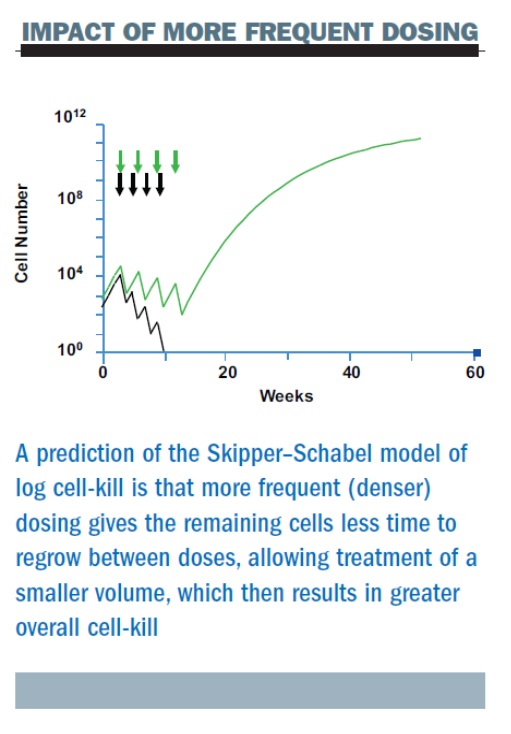 Is the log cell-kill model reflected in the clinic?
Is the log cell-kill model reflected in the clinic?
A study from Milan (see below) explored sequential or alternating treatment with CMF (cyclophosphamide, methotrexate, 5-fluorouracil) (in yellow) and doxorubicin (in red). The theory was that alternating these non-cross-resistant treatments would yield greater cell-kill. That is the arm represented by the top row of the figure. As a control, they administered the same four doses of the doxorubicin first, followed by the same total eight doses of CMF sequentially. Over the nine months of treatment, every patient on this study received the same four drugs, CMF and doxorubicin, with the same size doses of each drug and the same total dose of each drug. This emerges as an elegant test of dose density. The results speak for themselves, favouring the dose-dense regimen.
 Janice Gabrilove and colleagues, at my institution, first used growth factors – specifically granulocyte colony-stimulating factor (G-CSF), also known as filgrastim – to reduce neutropenia and associated morbidity due to chemotherapy in patients with bladder cancer. Although they gave full chemotherapy at a standard interval, all of the patients (100%) had full recovery of blood counts by day 14, and would have been able to receive planned chemotherapy, compared to only 29% of those not given G-CSF (NEJM 1988; 318:1414–22). As investigators were beginning to explore significant dose escalation, based on the hypothesis that the dose-response relationship was linear, we instead went in a different direction and began to explore dose density, meaning shortening of the intervals between treatments.
Janice Gabrilove and colleagues, at my institution, first used growth factors – specifically granulocyte colony-stimulating factor (G-CSF), also known as filgrastim – to reduce neutropenia and associated morbidity due to chemotherapy in patients with bladder cancer. Although they gave full chemotherapy at a standard interval, all of the patients (100%) had full recovery of blood counts by day 14, and would have been able to receive planned chemotherapy, compared to only 29% of those not given G-CSF (NEJM 1988; 318:1414–22). As investigators were beginning to explore significant dose escalation, based on the hypothesis that the dose-response relationship was linear, we instead went in a different direction and began to explore dose density, meaning shortening of the intervals between treatments.
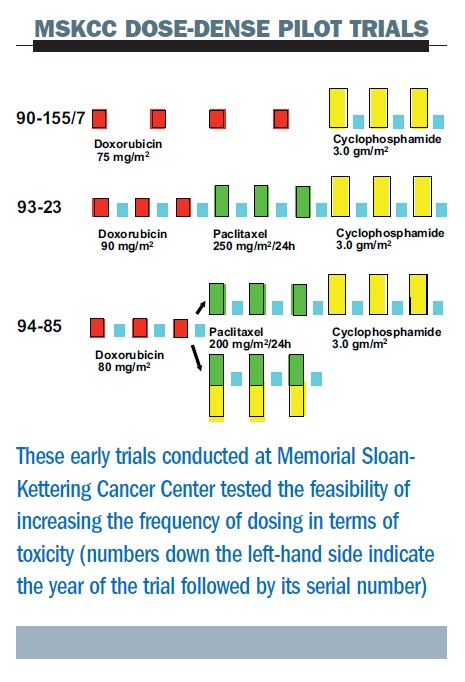
The figure below summarises three sequential pilot studies at the Memorial Sloan-Kettering Cancer Center (MSKCC). First, we were able to give high-dose cyclophosphamide (a very high dose of 3.0 g/m2) at two-week intervals with growth factor support. The second study added paclitaxel, in one of the first trials to add this, or any, taxane as adjuvant therapy (the ATC regimen – Adriamycin (doxorubicin), Taxol (paclitaxel), Cyclophosphamide). In this study the dose interval for doxorubicin was shortened – we gave three doses of each of the three drugs, all at two-week intervals and demonstrated feasibility, albeit with significant toxicities attributable to the use of higher doses of the individual agents than are currently employed.
Later, in a third study, we randomised patients to concurrent or sequential therapy with paclitaxel and cyclophosphamide, but all drugs were given in a dose-dense regimen. This study demonstrated that with these high doses, the concurrent regimen was no better in terms of toxicity. These studies were all too small (or non-randomised) to allow for efficacy comparisons.
 As one considers the results of trials that employ dose-dense regimens, it is important to be wary of possible confounders that can compromise the interpretation of such studies. For example, while we can achieve a dose-dense regimen with short intervals, testing it requires carefully controlled studies. Comparing four cycles of low-dose versus high-dose chemotherapy tests dose size. Comparing four cycles of a drug versus six cycles of the same size dose tests number of doses, and also tests total drug exposure, but not density. Controlling dose size but changing the frequency of administration – or density – while controlling the total dose number, is a pure test of dose density.
As one considers the results of trials that employ dose-dense regimens, it is important to be wary of possible confounders that can compromise the interpretation of such studies. For example, while we can achieve a dose-dense regimen with short intervals, testing it requires carefully controlled studies. Comparing four cycles of low-dose versus high-dose chemotherapy tests dose size. Comparing four cycles of a drug versus six cycles of the same size dose tests number of doses, and also tests total drug exposure, but not density. Controlling dose size but changing the frequency of administration – or density – while controlling the total dose number, is a pure test of dose density.
A typical design – and I have taken part in these studies myself – is four cycles of low-dose chemotherapy over three-week intervals, compared to three cycles of higher-dose every two weeks. This changes several parameters so it is not always clear what is being tested.
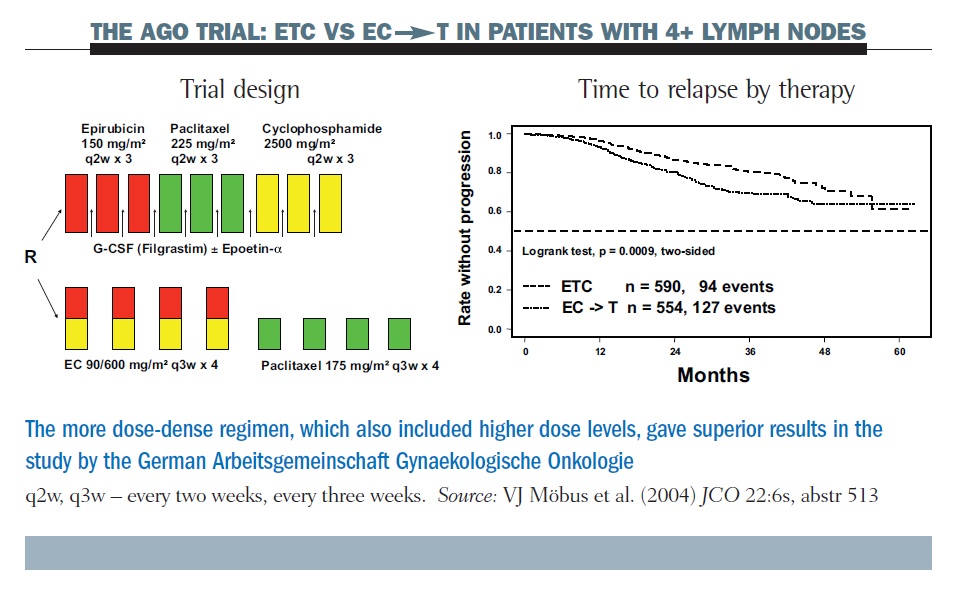
One example of a positive study was the AGO (Arbeitsgemeinschaft Gynaekologische Onkologie) trial (see below). Here, a dose-dense regimen of epirubicin, paclitaxel and cyclophosphamide (ETC), given with growth factor support, was superior in long-term follow-up to the conventional epirubicin/ cyclophosphamide (EC) paclitaxel regimen (JCO 2004, 22:6s, abstr 513). However, the number of doses of the three drugs varies and the size of the doses varies, as well as the dosing interval. Hence, while this study clearly demonstrated the superiority of a dose-dense regimen, critics could claim that this was due to other factors, such as the larger doses of the individual drugs.
 Weekly paclitaxel has been called “dose-dense” by us and others. However, here again there can be confusion in terms of what is tested in clinical trials. In the ECOG 1199 (Eastern Cooperative Oncology Group) study, AC was given at three-week intervals, followed by one of two taxanes – paclitaxel or docetaxel – using one of two schedules: weekly or three-weekly (q3).
Weekly paclitaxel has been called “dose-dense” by us and others. However, here again there can be confusion in terms of what is tested in clinical trials. In the ECOG 1199 (Eastern Cooperative Oncology Group) study, AC was given at three-week intervals, followed by one of two taxanes – paclitaxel or docetaxel – using one of two schedules: weekly or three-weekly (q3).
Weekly paclitaxel appeared to be superior to q3 paclitaxel. However, we note that 80 mg/m2 weekly of paclitaxel for 12 weeks is not the same as 175 mg/m2 q3, and so there are multiple variables at work here: dose number, dose size and frequency of administration.
The Cancer and Leukemia Group B dose-density trial CALGB 97-41 also employed a factorial design (see below). We asked two questions: the first question was about the frequency of administration, comparing q2 therapy with G-CSF support to q3; the second compared concurrent AC therapy with sequential therapy. What makes this study interpretable for us is that every patient had the same four doses of the same three drugs. All that varies across the four treatment assignments is concurrent or sequential dosing, and dose density.
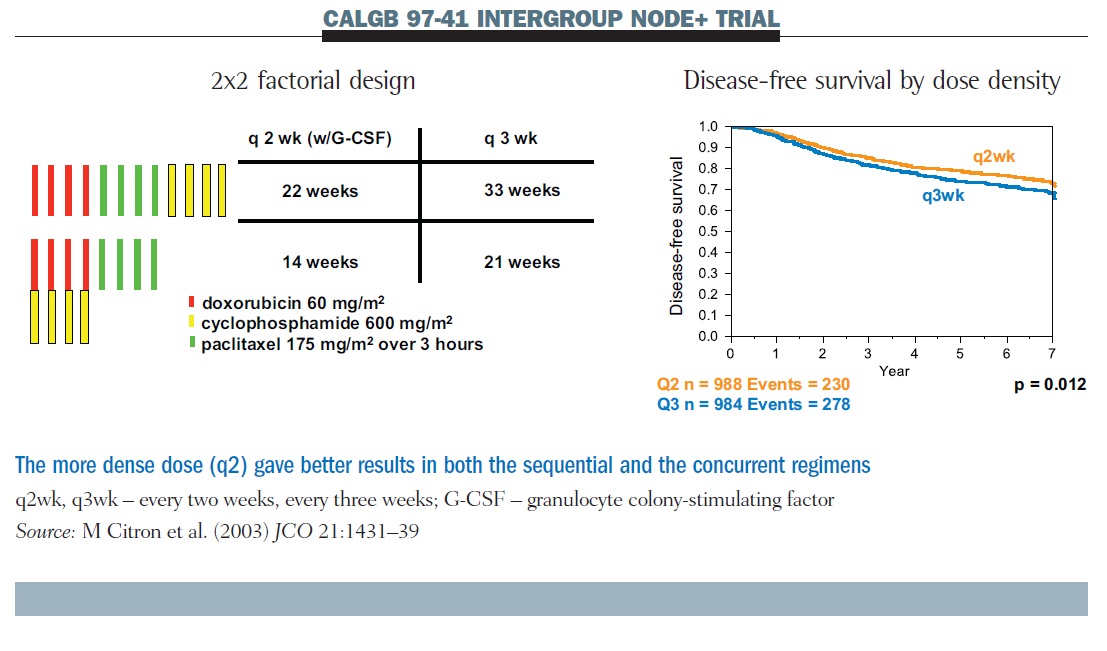 Results show that q2 therapy was superior to q3 for disease-free survival; this was also true for overall survival. There was no difference between sequential and concurrent therapy. We continue to use concurrent therapy most of the time because it allows us to get the treatment completed faster. But that is not the same as saying it is better, other than in terms of convenience.
Results show that q2 therapy was superior to q3 for disease-free survival; this was also true for overall survival. There was no difference between sequential and concurrent therapy. We continue to use concurrent therapy most of the time because it allows us to get the treatment completed faster. But that is not the same as saying it is better, other than in terms of convenience.
Toxicity
Once one accepts the superior efficacy of dose-dense treatment, the next concern is toxicity. This has become a particular issue in an era of trastuzumab and HER2-directed therapies for patients with HER2-positive disease.
The cardiotoxicity results from CALGB 97-41 showed the only acute cardiac event occurred in the patient you would have least expected: one treated with q3 single-agent doxorubicin. Looking at the total number of cardiac events – although this was purely exploratory and done retrospectively – showed that numerically there were twice as many events with q3 therapy as with q2 (2.5% vs 1.5%). This gave us some comfort that dose-dense therapy does not raise the risk of cardiac toxicity compared to q3.
This allowed us to go forward with pilot studies of dose-dense therapy and trastuzumab and also bevacizumab. The tables below show three studies of dose-dense AC with targeted therapy done by our group at MSKCC, and colleagues at the University of California San Francisco (UCSF), and the Dana-Farber Cancer Institute. The right-hand table summarises the cardiac toxicities, showing essentially no signal of acute cardiac toxicity over the four doses of AC across the several hundred patients.
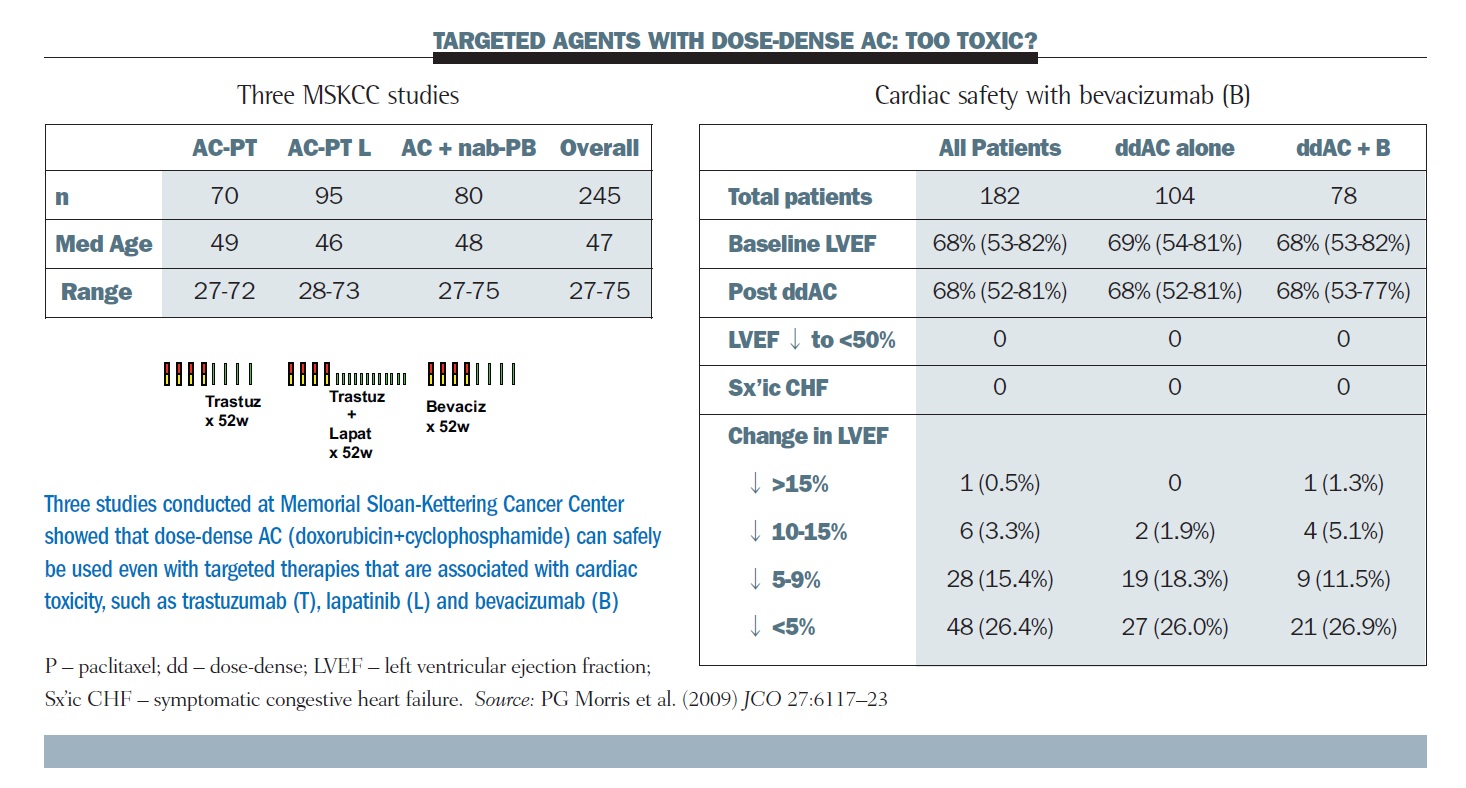 Longer term follow-up does not show any clear signal that dose density represents a special challenge for the delivery of full doses and durations of these regimens (JCO 2009, 27:6117–23). For comparison, in the cooperative group trials, about 65% of patients finished their full year of trastuzumab, whereas this number was about 80% in our studies.
Longer term follow-up does not show any clear signal that dose density represents a special challenge for the delivery of full doses and durations of these regimens (JCO 2009, 27:6117–23). For comparison, in the cooperative group trials, about 65% of patients finished their full year of trastuzumab, whereas this number was about 80% in our studies.
Taking dose-dense therapy forward
We incorporated the results of CALGB 97-41 into CALGB 40101. Initially this was a study of weekly paclitaxel for 12 or 18 weeks, versus AC q3 for four or six cycles with G-CSF. It was a two-by-two factorial design, comparing AC against single-agent paclitaxel for low-risk breast cancer. It was also a comparison of a longer therapy (six months) versus shorter (four months). There were those who argued that the superiority of AC followed by paclitaxel (or docetaxel) was not really attributable to taxanes per se but instead to the eight cycles of treatment which were presumed to be superior to four. Others have argued that six cycles of AC-containing therapy is better than four.
Based on the results of CALGB 97-41 we were motivated to change the study. With fewer than six hundred patients recruited, we modified it to include dose-dense therapy (q2 administration) and six cycles versus four. We continued the AC versus paclitaxel randomisation. In a still later modification of the study we dropped the six versus four cycle randomisation, making it a simple two-way comparison of paclitaxel versus AC, each then only administered for four cycles.
Results reported by Larry Shulman at San Antonio (2010) showed recurrence-free survival and overall survival with four cycles of treatment versus six were indistinguishable. We do not yet have the results of the AC versus paclitaxel comparison, but our data and safety monitoring board confirmed that they do not confound our results.
Is there a better AC or paclitaxel schedule?
SWOG study S0221 used a two-by-two factorial design of six cycles of dose-dense AC compared to a regimen of low-dose weekly doxorubicin regimen along with oral daily cyclophosphamide. Apart from that randomised comparison, they compared q2 paclitaxel for six cycles versus low-dose weekly paclitaxel for 12 weeks. Recently, they dropped the AC portion of the randomisation and shortened it to four cycles of every other week dosing. This was based on a futility analysis that weekly doxorubicin and oral cyclophosphamide could never be superior to the six cycles of AC. It does not mean it is worse. Hence the simplified design is now four doses of q2 AC, and the taxane comparison of low-dose weekly paclitaxel versus higher-dose q2 continues.
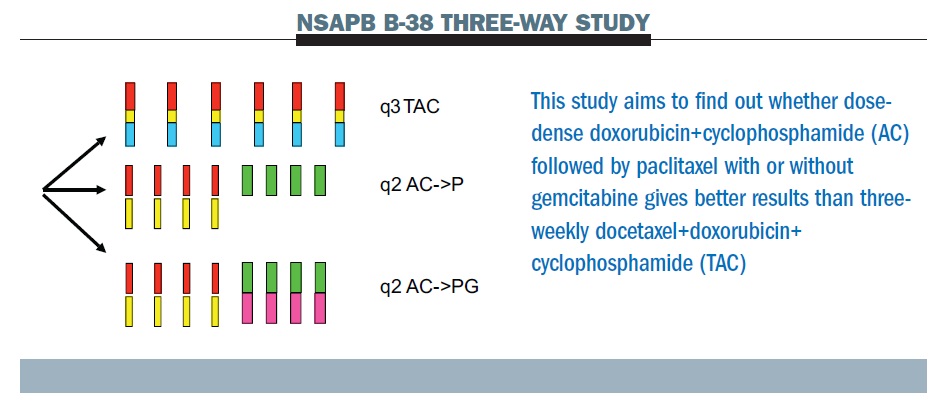
The NSAPB B-38 trial (see below) compares dose-dense AC paclitaxel (middle row) with TAC (top row – Taxotere [docetaxel], Adriamycin [doxorubicin], Cyclophosphamide) and experimental therapy of dose-dense AC paclitaxel with gemcitabine (bottom row). The tAnGo study, a UK-based trial that looked at the potential benefits of adding gemcitabine to an anthracycline- and taxane-containing adjuvant treatment regimen in early breast cancer, was negative. This suggests that the notion that gemcitabine as a fourth chemotherapy drug is going to add to this cohort of patients is unlikely to be supported.
 Can we further decrease intervals and increase dose density?
Can we further decrease intervals and increase dose density?
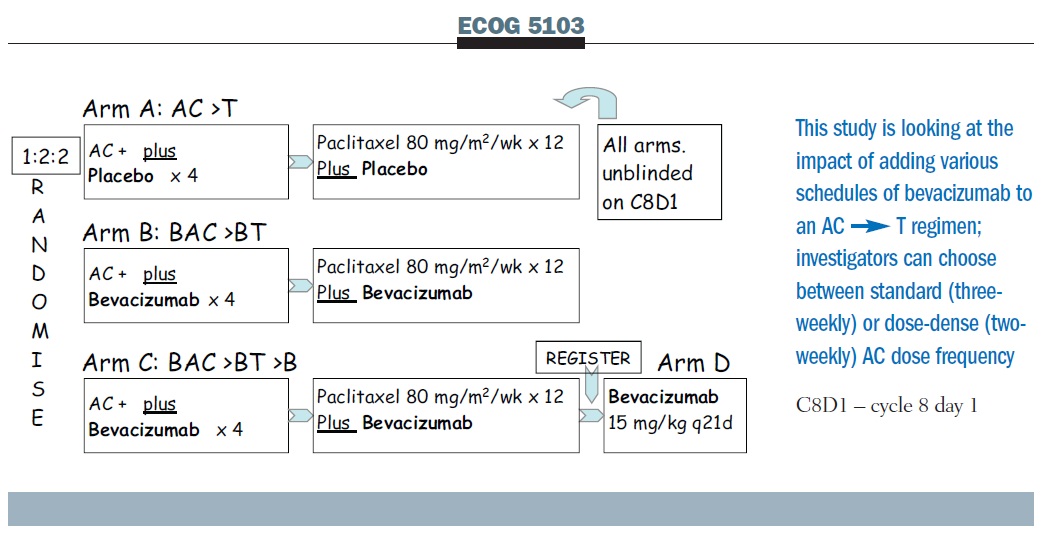
The ECOG 5103 bevacizumab trial is comparing AC, followed by weekly paclitaxel alone, with AC plus bevacizumab followed by paclitaxel plus bevacizumab, or AC followed by paclitaxel, both with bevacizumab and then followed by bevacizumab (see below). If bevacizumab adds a benefit, this study also allows us to ask about the duration of its use. Because clinicians have different views on the appropriateness of dose-dense therapy, the dose-dense regimen is allowed, as is q3 administration, and the patients were simply stratified on that basis.
 Our group has gone ahead asking whether we can push this further. The first pilot study, conducted by Monica Fornier, looked at 10- to 11-day intervals with sequential EC and paclitaxel using conventional G-CSF, because you cannot use pegylated G-CSF with such a short interval. The study demonstrated that this was feasible, but a randomised trial would be needed to show efficacy.
Our group has gone ahead asking whether we can push this further. The first pilot study, conducted by Monica Fornier, looked at 10- to 11-day intervals with sequential EC and paclitaxel using conventional G-CSF, because you cannot use pegylated G-CSF with such a short interval. The study demonstrated that this was feasible, but a randomised trial would be needed to show efficacy.
We then turned our attention to intravenous CMF, which was given two weeks on and two weeks off in the Milan studies in the past. Here we gave it every 14 days without breaks, which modelled the dose-dense experience of the CALGB. All we wanted to demonstrate was that it was feasible, because there are clinical reasons, from time to time in individual patients, to try to accelerate CMF, and when we treat patients in the low-risk setting this can be a viable alternative. For this not to be justifiable, we would have to show that shortening the interval makes the therapy less effective, but we have never seen evidence of that. Feasibility was strained at intervals of 10 –11 days but not at 14 days.
Dose-dense treatment in the palliative setting
Typically, we do not do studies of dose density in the palliative setting, because our goal here is not necessarily to achieve the highest response rate, or quickly deliver the lifetime tolerable (cardiac safe) dose of AC when it is used for palliation. Instead, our goal is to use the least toxic therapy that we can.
Capecitabine has high efficacy but also toxicity; giving the drug continuously for 14 days on a 21-day cycle results in a high rate of diarrhoea and gastrointestinal distress in the second week. We looked at mouse models of a capecitabine-sensitive tumour cell line. The maximal impact of therapy occurred eight days after starting treatment. This means that each day after that time point, if we continued to dose with capecitabine, cell-kill still occurred but it was less than the day before. The downside is that the toxicities accumulate so a week off is still needed to recover. We modelled the impact of a dose-dense schedule, which consists of one week on and one week off and this predicted that stopping therapy earlier, at one week, would allow for the earlier imposition of the needed seven-day break, but then an earlier re-initiation of treatment with resumption of greater cell-kill.
In the clinical extension of this work, our phase I study showed that this was feasible, and we have now done phase II studies with weekly (one week on, one week off) capecitabine combined with lapatinib or with bevacizumab, all of which have been feasible. This schedule has been widely adopted by clinicians because, as a practical matter, they so often have to stop before 14 days because of toxicity. This is a demonstration of the way in which a dose-dense schedule can be advantageous in the palliative setting as well as more curative in the adjuvant setting.
Conclusion
Dose scheduling – specifically in terms of density – is important, and it should be maintained in the adjuvant setting for both efficacy and toxicity. For example, using growth factor support with dose-dense AC not only enhances efficacy but also halves the hospitalisation rate (typically due to neutropenic fever). At the same time, it is fair to say that the cost issue is not fully addressed. Growth factor support is not inexpensive and the cost varies widely, making it unlikely that we will ever be able to develop an absolute answer on this issue. Cost-effectiveness in the curative setting depends, in part, on how much value is put on lives saved.
Finally, supportive care, in the form of growth factor use, is what facilitates the improved chemotherapy effect, so that is a critical part of the story. As we move further into the era of molecularly targeted therapies, it is important to note that dose-dense therapy does not preclude, and in fact supports, the use of these agents.
 Q: [Ukraine]: In your opinion, should we use metronomic chemotherapy or dose-dense chemotherapy? Which of the two will be the preferred option for the future?
Q: [Ukraine]: In your opinion, should we use metronomic chemotherapy or dose-dense chemotherapy? Which of the two will be the preferred option for the future?
CH: A metronome is, of course, the device that we use in piano lessons to keep time. The term is now being used, typically, to refer to low-dose weekly therapy, but essentially every regimen we ever use matches the metronome, with regular cycling of therapy. I reviewed a couple of studies with cyclophosphamide, doxorubicin and paclitaxel that directly answer the question on low-dose weekly therapy. Perhaps the best was a SWOG study with low-dose, weekly doxorubicin with oral daily cyclophosphamide compared to dose-dense AC, showing it was not better but somewhat more toxic. A study with low-dose weekly paclitaxel, which I suppose you could call metronomic, compared to q2 high-dose, or dose-dense, is open, so we do not have an answer. For other drugs, we would need to make comparisons to provide you with an evidence-based answer. That said, my heart lies with low-dose, less toxic therapy, especially in the palliative setting. I do not disagree with those who advocate metronomic chemotherapy as palliation for incurable disease, although I am a little less convinced that we have meaningful data yet in the adjuvant setting. Clearly, we, and others, are continuing to study this.
FC: I totally agree and I believe that we should probably test both, but my feeling is that what we call metronomic is probably better for the advanced setting, while dose-dense therapy makes more sense for the early setting. We need to let the trials end.
Q: Do you have any data about the long-term risk of leukaemia by adding G-CSF to dose-dense regimens for breast cancer?
CH: That was one of the interesting observations that we made – when we give AC across all of our CALGB studies, long-term follow-up averaged out at about a 0.5–0.7% incidence of acute myeloid leukaemia (AML). For our patient population, nearly half of those leukaemias are expected based on the natural history ageing rather than treatment. We have never demonstrated that growth factor support for a dose-dense regimen was associated with any increase in risk. For example, the incidence of AML in our study was 0.7% with q2 and q3 and was, paradoxically, higher with the sequential regimen in one of the comparisons and the concurrent regimen in the other. The NSAPB saw a significant increase of AML early on with dose-escalated cyclophosphamide and G-CSF support. When they gave 2400 mg/m2 of cyclophosphamide q3 with growth factor support, they saw an increased incidence, and I recall going to the National Cancer Institute in the 1990s to talk about whether this was worrisome. The problem here is that high-dose cyclophosphamide is clearly leukaemogenic. The dilemma is whether it is the growth factor causing this or the high-dose cyclophosphamide. In that context, CALGB 97-41 shows no difference in leukaemia with or without G-CSF. But where the doses are controlled and steady, I think it is probably not the case that G-CSF is contributing anything in terms of AML and lymphoma risk.
FC: If we look at non-dose-dense chemotherapy and the use of G-CSF in these situations, there is no conclusive evidence of an increased risk of leukaemia/ lymphoma in patients who need G-CSF, either as primary or secondary prophylaxis.
Q: What could be the role of dose-dense chemotherapy in the neoadjuvant setting?
CH: This question is not coming up quite as much these days, but used to come up quite a lot. Looking at the data, people are convinced of the benefit of giving dose-dense therapy postoperatively, but when I am trying to shrink a cancer preoperatively, I would give q3. This is because we do not yet have the right data to prove that a dose-dense regimen is better preoperatively.
The European School of Oncology presents weekly e-grandrounds which offer participants the opportunity to discuss a range of cutting-edge issues, from controversial areas and the latest scientific developments to challenging clinical cases, with leading European experts in the field. One of these is selected for publication in each issue of Cancer World. In this issue, Clifford Hudis, from Memorial Sloan-Kettering Cancer Center, New York, provides an update on optimising dose-dense regimens for women with early breast cancer. This is based on a News and Views article in Nature Reviews Clinical Oncology (2010, 7:678–679). Fatima Cardoso, from Champalimaud Cancer Centre, in Lisbon, Portugal, poses questions arising during the e-grandround live presentation. It as summarised by Susan Mayor. The recorded version of this and other e-grandrounds, together with 15 minutes of discussion, is available at www.e-eso.net