As patents on the first generation of monoclonal antibodies begin to expire, the cancer community will need to get to grips with the unique issues involved in ensuring the safety and efficacy of copies of these complex drugs made by living cells.
European patents have begun to expire on the first generation of biological anti-cancer drugs.
This should be good news for patients. Opening the market to competitors should help reduce the price tags of monoclonal antibodies, which should in turn give greater access to more patients. It should also generate savings that can be used to help pay for the new generation of biologicals that are now coming onto the market at even higher prices, including immunotherapies.
How far these benefits are realised in practice, however, depends not just on whether one or more competitor drug enters the market, and at what price, but the extent to which they replace the original drug in clinical practice.
Rituximab (MabThera) the first CD20 inhibitor, used in oncology to treat chronic lymphocytic leukaemia and some Non-Hodgkin lymphomas, came off patent in Europe more than two years ago. Cetuximab (Erbitux), the first biological EGFR blocker, used for certain advanced colorectal and head and neck cancers, came off patent in June 2014, closely followed by trastuzumab (Herceptin), the first HER2 blocker, which is approved to treat breast and metastatic gastric cancers overexpressing HER2. The European patent on bevacizumab (Avastin), the first angiogenesis inhibitor, will expire in 2022.
If cheaper copies are to be used in their place, doctors and patients will need to be confident that the competitor can be trusted to offer equivalent efficacy and safety. This raises the same issues of regulatory oversight that are associated with the approval and use of generics, but with an added twist: as biological drugs are derived from living cells, competitor drugs can never be exact copies of the original – which is why they are known as ‘biosimilars’ rather than ‘generics’.
Big money
Exactly how much cancer systems could save by switching to biosimilar monoclonal antibodies remains a matter of speculation, because none has yet been approved for the European oncology market. This is expected to change in the next year or two: Sandoz (a division of Novartis), Hospira (bought by Pfizer earlier this years) and Amgen all have anti-cancer biosimilars in the pipeline, as do a number of biologic drug specialists such as the Swiss company BioXpress and Polish company Mabion.
Biosimilars of an earlier, less complex, generation of biological drugs have, however, been routinely used in Europe for almost a decade now. These include the erythropoiesis-stimulating agents (ESAs) epoetin and the granulocytic colony-stimulating factor filgrastim, which are used in oncology as supportive care rather than anti-cancer therapies.
Some evidence is available to show their economic impact, both real and potential.
The German IGES Institute, for instance, has estimated that switching to a biosimilar ESA saved the country around €60 million in the first year. A study based on economic modeling, published last year (Future Oncol 10:1599–1609), estimated that potential savings from switching 100% to using a biosimilar ESA across five European countries – Germany, France, Italy, Spain and the UK – could be as high as €146 million, which the authors calculated would be enough to fund the treatment of up to 12,913 additional patients with MabThera, 5171 with Avastin or 4908 with Herceptin.
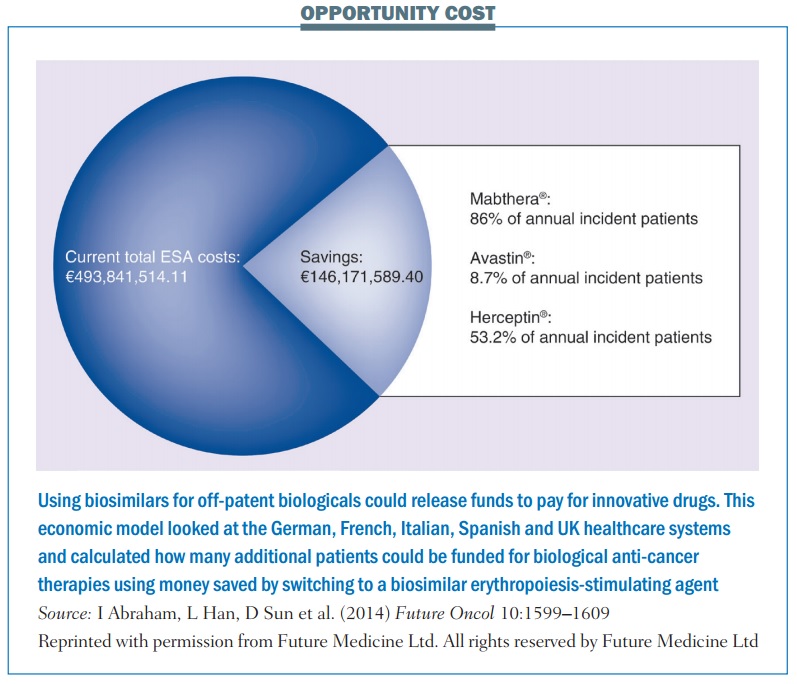
The question for the future will be how many more people could potentially get access to the latest immunotherapies – such as ipilimumab, nivolumab and pembrolizumab – and other innovative cancer therapies, if and when biosimilar versions of rituximab, trastuzumab and other off-patent monoclonal antibodies come on the European market.
The complexity of manufacturing monoclonal antibodies means that the price difference between the originator drug and any biosimilar is expected to be smaller in percentage terms than for generics and the first generation of biosimilars. But their high price means that even a small percentage reduction would yield significant savings.
Estimates from the PharmaNanoGene group at the University of the Basque Country’s Faculty of Pharmacy, suggest that introduction of biosimilar monoclonal antibodies could save more than €20 billion across Europe by 2020 (http://tinyurl.com/biosimsavings).
These eye-watering sums could give a welcome boost to efforts to sustain high-quality care for Europe’s growing number of cancer patients and survivors.
Achieving anything like that level of savings, however, would require a far greater take up by prescribers than has been seen with the first generation of biosimilars (see below).
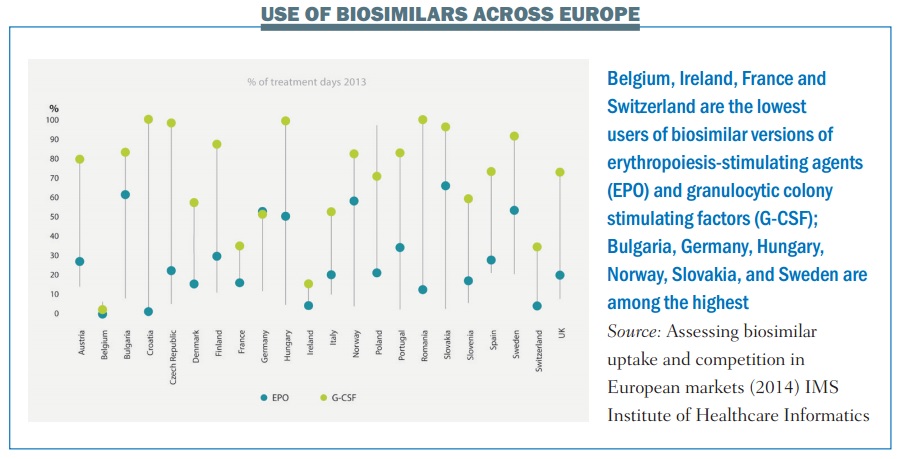
This won’t happen unless doctors and patients have confidence that any biosimilar approved for the European market is sufficiently similar to the ‘reference’ drug to be trusted.
How similar are biosimilars?
The regulatory challenge of assessing biosimilars for market approval was first flagged up in 2002 in an article titled ‘“Biogenerics”: the Off-patent Bio-tech Products’ (Trends PharmacolSci 23:119–121). Lead author HuubSchellekens, a professor of pharmaceutical biotechnology at Utrecht University, explains that the complexity arises because biological drugs are derived from living cells, which have natural variations: “Their size and complexity is on a different scale from other types of drug, giving more scope for variations.”
So while trastuzumab and lapatinib both target HER2, the former is almost 150 times heavier than the latter. Complexity of production is another factor, requiring close observation in highly controlled biotechnological labs, often taking many weeks. “It’s very difficult to keep the production conditions constant and homogeneous,” says Schellekens.
As biological processes are natural, random, and error-prone, the cells that express the monoclonal antibody protein cannot be identical, even in controlled conditions. As soon as cells make a protein, processes beyond the control of any biotechnologist take over.
Schellekens gives an example of the natural addition of sugar molecules to amino acids in proteins, known as glycosylation. “Differences in glycosylation patterns between two batches of the same biologic, for instance, could render them non-identical. Many such complications can modify a protein drug.”
Things can go wrong at any stage, from the start of the production process to pharmacist’s shelf. As cells spew out biologics, they undergo stresses from acid, heat and other ‘crowding proteins’ that the cells also produce, potentially leading to structural damage.
The physical and chemical milieu can also affect the next stage of production: extracting and purifying the biologics to obtain a homogeneous batch. Finally, the purified biologic is mixed with inactive ingredients to bring them to a final pill/injectable form. The added excipients, as well as the environment where the drugs are stored, could both act on the biologic drug. “Anything could go wrong anywhere in these steps,” says Schellekens.
But as he points out, these are issues not only for biosimilars, but equally for the original reference drugs, particularly as adjustments are often made to the production process. According to a paper from the Danish health authority, for instance, the manufacturers of infliximab (Remicade), a biologic used to treat conditions including rheumatoid arthritis and Crohn’s disease, made more than 35 changes to the production process over 14 years following approval in 1998. These included switching suppliers of cell culture media, moving production sites and introducing new purification steps.
Regulating biosimilars
The complexity of these molecules means that characterising differences between an original biologic and a biosimilar version can only be done through highly sophisticated bio-analytical technologies. The natural variations mean that it is difficult to draw firm conclusions about clinical comparability without seeing evidence that the biosimilar behaves in a comparable manner when used in actual patients.
In addition to quality data showing comparability with the production process of the original drug, and data on how the pharmacokinetics and pharmacodynamics compare, companies are therefore also required to show that their biosimilar demonstrates no clinically meaningful differences, either in efficacy or safety.
This will normally require randomised controlled trials, although not on the scale required for marketing approval of the original innovator drugs. These trials will be looking for equivalence, not superiority, and they will not measure ‘hard clinical endpoints’, such as survival. In the case of biosimilars for treating solid tumours, for instance, the EMA has indicated that overall tumour response would be a suitable endpoint to demonstrate comparable activity (www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/11/WC500099361.pdf).
There are pragmatic reasons for this. Requiring every biosimilar to jump the same hoops as an originator drug would be unethical, as patients risk losing if the biosimilar is less effective or safe, but have nothing to gain if equivalence is shown. The additional costs of running such trials would also greatly reduce the incentive to manufacturers and potential savings to health services. The regulators, both in Europe and the US, insist, however, that, there is also a strong scientific rationale for using endpoints that show a more immediate impact of the drug, arguing that they are less affected by patient- and disease-related factors than are endpoints such as progression-free and overall survival.
One of the big safety issues associated with all biological drugs is their propensity to stimulate an immune response as the body attacks what it identifies as a foreign invader. Such responses could render the drug ineffective – a big concern for patients with a life-threatening disease. They can also be dangerous, as demonstrated in Thailand, where an immune response to copies of epoetin, produced and marketed under the less stringent pharma regulations in that country, led patients to develop pure red-cell aplasia.
Regulators are therefore particularly insistent on seeing clinical data that shows comparability of the immunogenicity profiles. They also require the biosimilar producers to draw up a risk management plan (RMP), just as they would require for an originator. “The RMP for a biosimilar should take into account the identified and potential risks associated with the use of the reference [originator] product and should detail how these issues will be addressed in post-marketing follow-up,” an EMA spokesperson told Cancer World. The RMP should also include procedures for batch-by-batch quality control after any changes to the manufacturing process.
Such pharmacovigilance is vital in order to trace any clinical issues that arise with respect to both originator and biosimilar drugs, particularly to keep track of their immunogenicity profiles. Links to the periodic safety assessment reports for all products approved from February 2015 are available on the EMA website under the European public assessment reports (EPAR) webpage. RMP summaries for medicines authorised before this date will also be published when variations to their marketing authorisations result in significant changes to the RMP, said the spokesperson.
A question of confidence
Schellekens firmly believes that the regulatory procedures governing the approval of biosimilars for the European market are fit for purpose. A microbiologist by training, he has been a member of the Dutch Medicines Evaluation Board, a national expert of the European Medicine Agency, and a member of the Board of the European Immunogenicity Platform. He has published hundreds of papers in peer reviewed international journals, including on the immunogenicity of protein drugs and the problems related to biosimilars.
“Scientifically, biosimilars are already proven to be clinically safe and efficacious. Only political and bureaucratic caveats exist in communicating their true potential,” he argues. Indeed he believes that the current pharmacovigilance requirements, based on spontaneous reporting of adverse effects, may place an unnecessary burden on biosimilar producers, which could push up the price.
“Scientifically, biosimilars are already proven to be safe and effective”
He would prefer to see dedicated pharmacovigilance to look for defined effects such as immunogenicity, to narrow the time to trace back any issues. The money spent on expensive RMPs and EPARs could then be channeled into educating physicians and patients, he says.
While educating professionals, patients, policy makers and the public about the science and related regulatory issues will no doubt be important in building confidence and trust in biosimilars, that still leaves some outstanding concerns.
When is it OK to extrapolate?
The issue of extrapolation is a case in point. Can a biosimilar approved on the basis of clinical data from patients with one indication also be approved for use in other indications for which its reference drug is approved, without actually being tested in these other patient populations?
In an article published last year in Blood (vol 124, pp 3191–96) Martina Weise, vice-chair of the EMA’s Biosimilar Medicinal Products Working Party, explained the EMA’s approach to extrapolation: “If a biosimilar producer establishes the relevant mechanism of action of the biologic and its target in the human body, such as a receptor that receives the biologic, then extrapolation is usually not problematic.” Each case needs to be carefully considered on its own merits, however, an EMA spokesperson told Cancer World, “and therefore the possibility for extrapolation is limited and needs to be fully justified.”
The EMA’s judgement on what is justified has been questioned, however, by the European Crohn’s and Colitis Organisation, which is unhappy with the 2013 EMA decision to include Crohn’s disease and ulcerative colitis among the indications for which Rensima, a biosimilar version of the anti-inflammatory biologic Remicade (infliximab) was approved. Rensima had shown clinical comparability with Remicade in reducing the rate of progression of joint damage in patients with rheumatoid arthritis, and was subsequently approved for treating patients with rheumatoid arthritis, ulcerative colitis, ankylosingspondylitis, psoriatic arthritis and Crohn’s disease.
The EMA’s judgement has been questioned by the European Crohn’s and Colitis Organisation
The EMA clearly felt that extrapolation from the clinical studies done in rheumatoid patients was justified. However, in a position statement published on the Genetics and Biosimilars Initiative website (www.gabionline.net), the European Crohn’s and Colitis Organisation asserted that: “… the use of biosimilars in IBD patients [patients with inflammatory bowel disease – i.e. Crohn’s disease or colitis] will require testing in this population with comparison to the appropriate originator product. Clinical efficacy in IBD cannot be predicted by effectiveness in other indications such as rheumatoid arthritis.”
Last year the Canadian regulator decided against using clinical data from patients with rheumatoid arthritis as a basis for approving Rensima for treating Crohn’s disease or ulcerative colitis, “due to differences between Rensima and the reference product that could have an impact on the clinical safety and efficacy of these products in these indications,” though it did approve the biosimilar for ankylosingspondylitis, psoriatic arthritis and psoriasis. The US regulators have currently postponed their decision, pending further information.
Similar controversy could potentially arise with the first rituximabbiosimilars, over whether demonstrating clinical comparability in shrinking lymphoma tumours can be extrapolated to indicate comparability in treating rheumatoid arthritis. Schellekens points out that unless there is a strong scientific rationale for a separate trial, the added development costs would push up the price for the rituximabbiosimilar with no good reason.
Are biosimilars interchangeable?
Another area where doctors and patients may seek reassurance is over whether biosimilars can be considered interchangeable with the originator drug. Would doctors be able – or be required – to switch patients from the original drug to its biosimilar, be able to switch patients back again, or even switch between biosimilars?
Would pharmacists – or patients – have the right to substitute the biosimilar if the prescription was for the originator drug? Karen Van Rassel, CEO of the patient advocacy group Lymphoma Coalition, says patients are still looking for clarification.
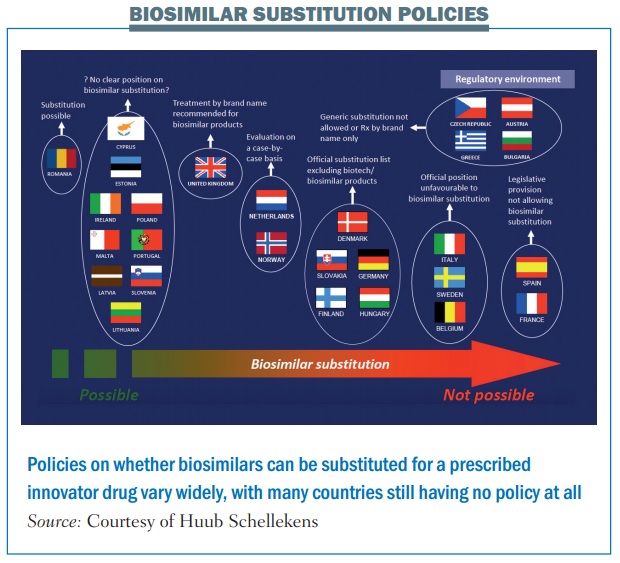
According to PekkaKurki, at the Finnish Medicines Agency (Fimea), it is not within the EMA’s remit to provide that clarification. Addressing a joint meeting of Patients’ and Consumers’ Organisations (PCWP) and Healthcare Professionals’ Organisations (HCPWP) at the EMA, earlier this year, he said “EMA thinks it does not have the mandate on interchangeability because it comes very close to substitution, which is a national issue.”
The problem is, as he adds, that very few national bodies have issued official recommendations on the interchangeability of biosimilars (see figure).
Matti Aapro, Dean of the Multidisciplinary Oncology Institute, in Genolier, Switzerland, has been studying and administering biosimilars for many years, in his capacity as a medical oncologist specialising in supportive cancer care. He argues that there are no grounds to believe interchanging between innovator products and biosimilars could create problems. “There is no scientific evidence at all that there is a biological or clinical risk if you change from one product to another,” he says. He agrees, however, that if a patient or a pharmacist wishes to interchange between an originator product and a biosimilar, or between biosimilars, for reasons of cost, they should do so with the knowledge of the prescribing physician. “Physicians should keep stringent records on any interchange, switch, or substitution, so that if any problem occurs due to this change, the issue can be traced back.”
Transparency and the battle for perceptions
The principle of transparency – that professionals and patients should always know whether a given drug is the original or a biosimilar – is widely recognised as a cornerstone of building confidence in biosimilars. Exactly how that should be reflected in the way they are named, however, remains a matter of controversy.
There is agreement that each biosimilar should have its own brand name or unique identifier, but should it be allowed to share the same ‘generic’ or ‘international non-proprietary name’ (INN) as the innovator product? Should a biosimilar of Herceptin go under the INN ‘trastuzumab’, or should its INN carry a ‘biological qualifier’ eg a suffix that indicates the drug is a biosimilar, or identifies the company that made it? Arguments centre on the potential for confusion and error.
Adding a suffix, some argue, will reduce the likelihood of inadvertent and inappropriate product switching and strengthen the accuracy of tracing via post-marketing safety monitoring systems. Others suggest, however, that the system of brand name plus INN is a worldwide system that works well, and adding a layer of complexity will actually increase the chance of errors.
While these concerns are legitimate, they also act as a surrogate for a battle over perceptions. Adding a qualifier to the INN sends out a signal that the biosimilar is not really a version of the same drug. Using the same INN conveys the opposite. Perceptions are likely to have a significant impact on the extent to which biosimilars are adopted into clinical practice.
In August 2015, the FDA published its long-awaited guidelines on the naming of biosimilars, coming out in favour of a suffix.
The recently approved biosimilar for Amgen’s Neupogen – INN “filgrastim” – has accordingly been given the INN “filgrastim-sndz”, to indicate it is a biosimilar manufactured by Sandoz.
Europe, in contrast, seems to be leaning towards using identical INNs. Concern that a different INN for biosimilars “could undermine the trust of healthcare professionals and the public” was one reason put forward in favour of this position, according to minutes from the October 2013 meeting of European Commission’s Pharmaceutical Committee.
This is not unreasonable, given the €20 billion that some projections estimate could be saved across Europe by 2020 with a 100% switch to biosimilar monoclonal antibodies. But if Europe is going to achieve even a 50% switch rate, policy makers will not be able to rely on identical INNs to command public and professional confidence. They will need to build that trust not just through education, but also by listening to the concerns of the cancer community, and convincing them that the EMA approach to the regulatory challenge posed by biosimilars is scientifically sound.
PRINCIPLES OF APPROACH FOR BIOSIMILARS
EMA will require:
- Full quality dossier (covering the chemistry, manufacturing process and controls), including comparisons with original
- Limited preclinical dossier, including pharmacokinetic comparison with original n Clinical similarity – hard clinical endpoint not needed
- Extrapolation must be demonstrated from one condition to another
Risk management plan with post-marketing safety studies, including immunogenicity
Source: Adapted from the EMA Guideline on Similar Biological Medicinal