Dose too strongly and the patient gets more harm than benefit, too weakly and the drug cant do its job. Could oncology learn from other fields about using pharmacokinetics in the clinic to hit it right in each patient?
 Personalised medicine is about tailoring treatment and care to the individual patient and their specific disease. However, oncology has so far largely resisted the idea of personalising dose levels, despite what is known about wide variations in individual pharmacokinetics, which govern how patients’ bodies absorb, metabolise, distribute and clear therapeutic drugs.
Personalised medicine is about tailoring treatment and care to the individual patient and their specific disease. However, oncology has so far largely resisted the idea of personalising dose levels, despite what is known about wide variations in individual pharmacokinetics, which govern how patients’ bodies absorb, metabolise, distribute and clear therapeutic drugs.
Conventionally, dosage of anti-cancer drugs has been calculated according to the patient’s body surface, which can be estimated by weight and height or more simply by weight alone.
Leading pharmacologists, such as Silvio Garattini of the Mario Negri Institute in Italy, have been arguing for some time that oncologists need to pay more attention to pharmacokinetics (eg EJC 2007, 43:271–282). Poor responses – or indeed unexpectedly severe side-effects – they argue could be the result of a conventional approach to dosing that fails to take this into account.
One consequence, they say, is that patients may be wrongly taken off drugs that could benefit them, if used at the optimal dose. Another is that potentially valuable experimental drugs could be wrongly discarded for lack of efficacy or too high toxicity.
Calls are now growing for oncologists to monitor the drug levels circulating in the body as an essential element of personalising treatments.
Therapeutic drug monitoring (TDM) is already commonly used with a number of agents, including anticonvulsants for epilepsy, anti-coagulants such as warfarin, drugs that treat arrhythmia (cardiac disorder), lithium, some antibiotics, and immunosuppressants.
It is usually done by measuring drug concentrations in blood samples, with the timing and analysis of these tests done to suit the particular behaviour of a drug and its administration.
The main criteria for its use are where the drug has a narrow therapeutic window (i.e. a narrow dose range for which benefits outweigh risks) and a significant variability among patients in pharmacokinetics. There should also be a strong correlation between blood levels and therapeutic effect (ideally oncologists would like to know the drug levels in tumours, but that’s much more ambitious), and of course suitable tests and facilities must be available.
Another criterion could be where there is a reason for monitoring variations in dose within a single patient over time, for instance to help patients with adherence when the drug is taken over long time periods. Chronic myeloid leukaemia is the obvious example, and indeed imatinib (Glivec) is so far the only targeted drug for which the routine clinical use of therapeutic drug monitoring has been suggested.
Limited clinical use
Until now, clinical application of therapeutic drug monitoring in cancer patients has been limited to cytotoxics, including methotrexate (given for cancers such as acute lymphoblastic leukaemia) and 5-fluorouracil (given for common cancers such as colorectal and head and neck, and the object of study in one of the few major controlled trials of therapeutic drug monitoring). It is also used in treating children with highly toxic agents such a carboplatin.
Lately, however, there has been a revival of interest in the potential for greater use of therapeutic drug monitoring in cancer, sparked by a pan-European group of pharmacologists convened by the French Society of Oncology Pharmacy. Last August, for instance, the European Journal of Cancer ran a series of position papers on the issue, with the lead paper posing the question: ‘Therapeutic drug monitoring in cancer – are we missing a trick?’ (EJC 2014, 50:2005–09).
While pharmacologists may lack experience in introducing new techniques into clinical practice, they do have the specialist knowledge – and they make a strong case.
Jan Beumer, a pharmacologist at the University of Pittsburgh Cancer Institute, wrote a provocatively titled paper last year, ‘Without therapeutic drug monitoring, there is no personalised care’ (Nature Clin Pharm Ther 2013, 93:228–230). He says that pharmacokinetics – a longstanding science – has become marginalised today amid the boom in interest in and funding for all the ‘omics’ in oncology. “Clearly genetics has a role – without certain mutations, such as BRAF in melanoma, you know a drug won’t work. But drug exposure is the end result of a lot of variables, and blood concentration can be much more informative.”
He mentions 5-fluorouracil (5-FU) as a good example. Not only does it have the prerequisite narrow therapeutic window, but its pharmacokinetics also vary widely between patients. Most of this variability is the result of the activity of an enzyme, DPD, which clears 5-FU and if low can lead to a toxic build up that can be fatal. There is a test for the gene mutations that cause DPD deficiency, but most of the people who have severe side-effects have a negative result, says Beumer. Therapeutic drug monitoring, he argues, offers a way to address this variability (without having to explain it).
A randomised trial of therapeutic drug monitoring, currently the landmark trial for its use in oncology, was carried out on 5-FU by medical oncologist Erick Gamelin and colleagues in France, and published in 2008 in the Journal of Clinical Oncology (JCO 2008, 26:2099–2105). Ten years earlier they had found that, using standard dosing, a startling 43% of patients were not given the right dose – 33% being underdosed and 10% overdosed. Given that several studies had also shown a relationship between blood levels of 5-FU and the therapeutic window, they divided more than 200 patients into two groups – one receiving a standard dose and another a dose individually adjusted according to the pharmacokinetics. The results showed a significantly improved response rate, a trend to a higher survival rate, and fewer severe toxicities.
Ron Mathijssen, who is both a medical oncologist and pharmacologist at Erasmus Medical Centre in Rotterdam, and professor of individualised oncologic pharmacotherapy, says much of the new enthusiasm for therapeutic drug monitoring comes from understanding the role played by drug concentrations, thresholds of activity, and also huge variability among patients.
“For many years we just didn’t know about these factors,” he says, noting that there are many physiological factors that affect exposure apart from genetics, especially with oral targeted drugs. These could include age, gender, liver and kidney function, interactions with other drugs and foods, smoking, and differences in drug absorption with oral agents. A high-fat meal alone can give a large exposure effect on a drug such as lapatinib for breast cancer, says Mathijssen.
“Age, gender, liver and kidney function, interactions with other drugs and foods and smoking are all factors”
Because so many factors play a role there are few biomarkers that can predict drug exposure in cancer, unlike some other conditions (such as cholesterol levels for statins). “Kidney function is one which we use in drugs such as carboplatin, and the only option we have in current practice,” says Mathijssen. “There have been attempts to use genotyping for certain enzymes to see whether they lead to variations in concentrations, but in practice it can be too complicated. If there is variation in an enzyme (such as UGT1A1*28 for irinotecan), a medical oncologist is more likely to switch to another drug that doesn’t have that enzyme variation, which is unfortunate as it means we are not using drugs in an optimal way.”
Dose calculations
In the absence of therapeutic drug monitoring or other biomarkers, for cytotoxics, oncologists rely on dose calculations based on body surface area (BSA), as used in the control arm of the 5-FU study. And going further back to drug development, dosing is set in phase I studies and fine-tuned in phase II, says Beumer, by setting the maximum tolerated dose (MTD) for the whole population from only a few patients. “Just one in six patients at phase I who get toxicity set the MTD,” he says.
The starting doses for the trials come from interspecies extrapolation of BSA – from mice up to humans – which scales well with metabolic capacity. “But it doesn’t work within a species,” says Beumer. “There’s an assumption that someone with a bigger liver metabolises more so can receive a bigger dose, but in fact a small 60-year-old woman may have much more metabolic activity than me, a much younger man well over six feet tall.” Beumer and colleagues have written that BSA-based dosing “gives the false impression that we are practising personalised medicine by using a patient-specific metric.”
This, he says, has resulted in several drugs such as paclitaxel and capecitabine being approved at doses that are too high for most people, and oncologists know to take the dose down. Both he and Mathijssen comment that oncologists obviously like their patients to feel good during treatment. “Where there is toxicity we lower the dose say by 25% or 50%, but we never upgrade the dose if there is no toxicity say by trying 50% more,” says Mathijssen. “Wearing my pharmacologist’s hat I don’t think this is right, but it is hard to change as many medical oncologists like it when the patient has no complaints. The result though is that we are underdosing almost all our patients, in a certain way.”
“Where there is toxicity we lower the dose, but we never upgrade the dose if there is no toxicity”
Though, as Beumer points out, if a patient on a drug such as 5-FU does suffer toxic side-effects, even reducing the dose does not necessarily mean that the drug becomes tolerable, and patients can drop out of treatment and lose a chance to respond. “We have tried dosing up to toxicity in trials but it has not been successful,” he adds. “Then if you go too low people tolerate it, but the tumour progresses and you go to the next line, which may mean using more expensive drugs.”
Practicalities
There are a number of barriers and objections to using therapeutic drug monitoring in oncology. Jaap Verweij, a medical oncologist and now dean and vice-chair at Erasmus MC in Rotterdam, argues that practical difficulties mean it’s currently hard to apply in clinical practice. “While we can do beautiful studies in an academic setting, where we can get an almost immediate answer to the questions we ask, these facilities just aren’t available to most clinicians, and if you wait several days for test results that’s a big limitation.”
Organising patients for testing can also be hard, says Beumer: “My oncologist colleagues say therapeutic drug monitoring is great, but when they try to implement it they find that a patient may live two hours away, and it would be hard to get them to come in for an extra blood draw say for 5-FU.”
“It would be hard to get them to come in for an extra blood draw say for 5-FU”
Cost is also an issue, and there are also regulatory barriers, which may be one reason pharmaceutical companies are not promoting its use. Beumer points to a warning letter sent in 2010 from the US regulator, the FDA, to Novartis about claims made on company-sponsored sites about the use of therapeutic drug monitoring with imatinib, which “urged physicians to measure the plasma concentration of tyrosine kinase inhibitor in their patients’ blood, and then use that information to individualise the drug’s dosage or schedule.”
The FDA said it was not aware of “substantial evidence or substantial clinical experience to support a correlation between patient outcome and plasma levels of imatinib,” and that the prescribing information has no provision for monitoring and increasing drug doses, only reduction or discontinuation for adverse events.
Building an evidence base
More evidence has arrived since then, but to get therapeutic drug monitoring accepted for widespread clinical practice it is likely that prospective randomized trials will be needed to validate significant effects that are seen in current retrospective work. Funding and interest for such trials, however, may be hard to come by, especially for older off-patent chemotherapy drugs.
“Prospective randomised trials will be needed to validate the significant effects seen in retrospective work”
Trials are more likely in oral targeted agents, not least because of their sheer cost – healthcare funders may well ask more questions about whether these expensive drugs are being efficiently deployed. Verweij notes that exposure studies after first doses in a drug such as the immune therapy ipilimumab for melanoma could save a huge amount of money by identifying earlier who are the 20% of patients who respond.
In their review of therapeutic drug monitoring of targeted therapies in the European Journal of Cancer (EJC 2014, 2020–36), the pan-European group note that it could provide additional information on efficacy, adherence and safety compared with clinical evaluation alone. Most studies so far have focused on TKIs, which in the case of imatinib have led to clinical recommendations, with the European Society for Medical Oncology currently suggesting that therapeutic drug monitoring may be important in all patients and is recommended for some, such as those who have suboptimal response.
Mathijssen notes that imatinib has a fairly well-established threshold of activity that lends itself to such recommendations – studies look at the ‘trough’ concentrations of the TKIs, which are easier to assess than the more intensive monitoring needed for cytotoxic drugs.
Verweij is doubtful about the clinical applicability of therapeutic drug monitoring at least for chemotherapy, and says it is certainly of limited use for 5-FU. “5-FU has a very short half-life so you need a test close to the bedside, it is very cheap so economic benefit is low, and its efficacy is limited. And it’s hardly ever used on its own, so there is the added complexity of what a patient may be responding to,” (though Mathijssen says it is still important to set optimal doses in combinations).
Michèle Boisdron-Celle, a pharmacologist colleague of Erick Gamelin, disagrees, saying that, given the right tools, therapeutic drug monitoring is relatively easy and not costly. They have set up a company in France, ODPM, which provides calculators for predicting the risk of toxicity of the fluoropyrimidine family of drugs (5-FU, capecitabine, etc) and for dose adaptation, which they consider give clinicians tools for better managing patients – not least to avoid deaths caused by toxicity.
“The literature on TDM is conclusive as far as we are concerned,” says Boisdron-Celle, and while there “may be some data missing in terms of randomised controlled studies, if there is a clinically proven method for avoiding risks there is a serious ethical question that is raised by asking for RCTs.”
In Rotterdam, Mathijssen’s group is doing feasibility trials for therapeutic drug monitoring trials on other TKIs such as sunitinib and pazopanib, and is carrying out drug concentration work for a new trial in Switzerland, Austria and Germany on cabazitaxel, a new taxane chemotherapy for prostate cancer. Tamoxifen, an old targeted agent for breast cancer, is also a good candidate, he notes.
There is much exasperation about the lack of interest and understanding of therapeutic drug monitoring among healthcare providers, oncologists and drug companies. Boisdron-Celle is particularly concerned by the pharmaceutical industry. “The benefits of TDM have been clear to all pharmacobiologists and pharmacogeneticists for over 20 years. It’s simply easier for the industry to give a standard dose to everyone,” she says.
In the US, an equally forceful commercial voice is Salvatore Salmone, who runs a company called Saladax that currently offers assays for 5-FU, docetaxel and paclitaxel, and who points to solid data in the pharmokinetic literature that tends to pass oncologists by.
“The benefits of TDM have been clear to all pharmaco-biologists and pharmacogeneticists for over 20 years”
Beumer, who has done some work with Saladax, says sessions on therapeutic drug monitoring at major cancer conferences tend to get sidelined. He hopes the International Congress of Therapeutic Drug Monitoring and Clinical Toxicology (ICTDMCT), a small organisation for which he helped found an oncology section, will contribute to improving the situation.
Mathijssen expects progress as tests and protocols become better suited to clinical practice. “It will be part of practice in ten years’ time I believe, because by then it should be possible to measure just one sample with cheaper assays at nearby facilities. I really think it’s important – although we don’t have the evidence of where the exact threshold might be for a lot of drugs, if we do measure a concentration below the lower limit of quantification we would know the drug is below the therapeutic range and is therefore inactive.”
AWARD-WINNING SERVICE FOR CHILDREN
Gareth Veal, co-author of the European Journal of Cancer series on therapeutic drug monitoring (2014, vol 50, pp 2005–09) and head of pharmacology research at the Northern Institute for Cancer Research, Newcastle, UK, specialises in both adult and paediatric pharmacology and has led the introduction of an award-winning therapeutic drug monitoring service for children treated with carboplatin. As he says, although a lot of drugs used with children have a good response rate they can be very toxic and difficult to manage in small children with developing organs. Many protocols therefore try to maintain response whilst minimising the side-effects of treatment, which is where therapeutic drug monitoring approaches can have an impact.
“In the UK we now routinely carry out carboplatin therapeutic drug monitoring from our reference centre in Newcastle, based on datasets we’ve built up from monitoring treatments over a number of years and from well-designed clinical trials. Two or three blood samples taken two hours after administration are sent to us and we have the results on dosing back to oncologists the next morning. We do it for groups where there is a high risk of toxicity or lack of response, and for neonates and infants, where drug disposition can be more difficult to predict.”
Veal says the carboplatin service is the only one he is aware of in Europe for this drug, but centres with similar expertise are working on other important drugs in children, such as busulfan at Gustave Roussy in Paris.
THE CASE FOR THERAPEUTIC DRUG MONITORING OF TKIs IN CML
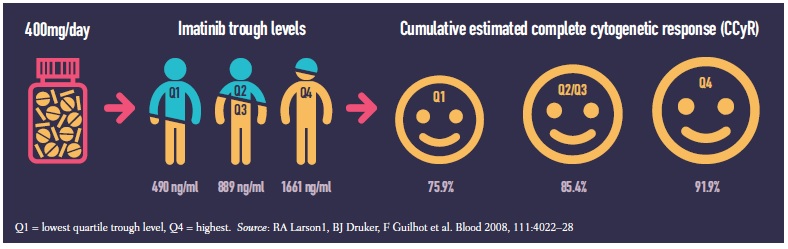
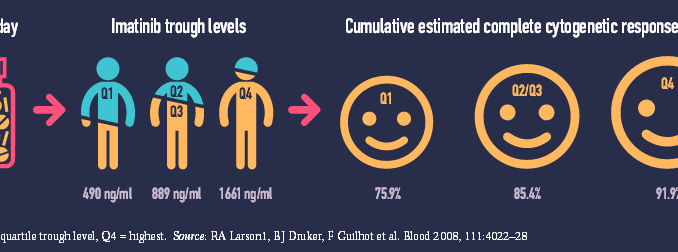
A 2008 study (Blood 2008, 111:4022–28) showed a 25-fold variation in trough levels of plasma concentration of imatinib (Glivec) in 351 patients treated for chronic myeloid leukaemia (range 153–3910 ng/ml). Patients who achieved the best possible outcome of complete cytogenetic response had significantly higher trough levels than those who did not (P=0.01). Patients with high imatinib exposure also had better rates of major molecular response and event-free survival.
While some serious (grade 3/4) adverse events increased at higher imatinib exposures others decreased. Over a five-year period, serious fatigue, fluid retention, myalgia and anaemia were all more common in patients in the highest exposure quartile (Q4) than the lowest (Q1), but patients with the lowest exposure suffered more serious joint pain, haemorrhage, rash, neutropenia and thrombocytopenia.
The study was a retrospective subanalysis of the IRIS trial (NEJM 2003, 348:994–1004), which five years earlier had compared imatinib against interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukaemia.
The US drug regulator, the FDA, has warned manufacturer Novartis against advising doctors to monitor imatinib plasma levels and adjust the dose accordingly, on the grounds that it was not aware of “substantial evidence or substantial clinical experience to support a correlation between patient outcome and plasma levels of imatinib”. Incorporate these pharmacokinetic studies in the pivotal trial design seems to be the message from the FDA.