Showing that a new drug can keep advanced cancers from progressing, or stop early cancers from returning, is quicker, cheaper and easier than showing that it helps patients live longer. But how can we judge in which instances these surrogates will accurately predict overall survival?
 Overall survival is the gold standard and primary outcome of interest for cancer clinical trials. It is an ‘appropriate measure’ for evaluating cancer drugs and therapies, based on recommendations from regulatory bodies who have declared that a primary outcome in clinical trials should demonstrate that a new treatment has some sort of clinical benefit (FDA, 2007).
Overall survival is the gold standard and primary outcome of interest for cancer clinical trials. It is an ‘appropriate measure’ for evaluating cancer drugs and therapies, based on recommendations from regulatory bodies who have declared that a primary outcome in clinical trials should demonstrate that a new treatment has some sort of clinical benefit (FDA, 2007).
In performing a clinical trial, there is an idea that all researchers need to show is an improvement in overall survival for a drug to be approved. It is of course not that simple. There are issues in following patients over longer follow-up periods, when there may be potential confounding with secondary or tertiary treatments making it hard to show which treatment contributed what to the overall survival.
In terms of clinical trials, the good news in cancer is that patients are living longer with therapies that are much more effective than 20 or 30 years ago. However, in terms of clinical trial design, there is increased risk of confounding factors with this longer life span. Patients will go on to get further line treatments than they would previously have been given. Additionally, because the patients are on-study for longer, trials also have to go on for longer, which increases costs for trials.
Early biomarkers
One of the questions for trial designers is whether we can identify some sort of early biomarker to use instead of overall survival that will give us an indication that a treatment is potentially effective. We ideally want a marker that requires a short period of time until the event occurs. A successful early biomarker will, most of the time, show a larger treatment effect than what we might see if we use overall survival (OS), and there should be less confounding, as patients receive fewer second- and third-line treatments. All of this will reduce the sample size required, reduce the length of time required for the clinical trial and, ultimately, reduce the cost of performing a clinical trial.
An example of a trial that used an early biomarker is the National Cancer Institute of Cancer MA.17 phase III randomised, controlled trial of letrozole in postmenopausal women with breast cancer who had previously completed five years of tamoxifen. The primary endpoint was disease-free survival (DFS). More than 5000 women were enrolled and at the first interim analysis, which occurred 2.4 years after the start of the trial, there were 207 DFS events (i.e. 207 women were no longer disease free).
The DFS plot (see left-hand graph, below) shows there is a separation of the curves between the letrozole group and the placebo group. The OS plot (right-hand graph) shows no separation between the two groups (NEJM 2003, 349:1793–1802). However, because of the difference in the DFS curves, the Data and Safety Monitoring Committee recommended stopping the trial on the grounds that letrozole had been shown to be superior to placebo.
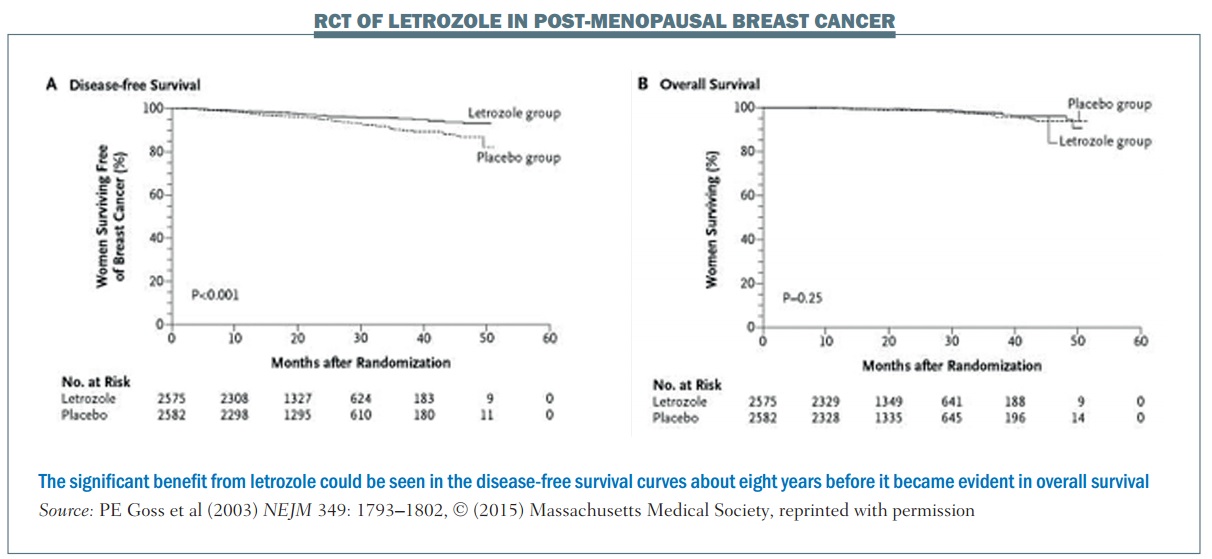
Long-term follow-up demonstrated improved overall survival with letrozole in this patient population. However, using OS rather than DFS to achieve the same level of significance (alpha=0.05) would require follow-up of about 10 years. Using the early biomarker of DFS meant that publication occurred about eight years earlier than it otherwise would have done. This is a good example of where using an earlier biomarker showed a great advantage over OS, enabling earlier publication showing the same statistically significant results.
There are two ways we can find early biomarkers to improve clinical trial efficiency:
- Find a marker that shows clinical benefit
- Find some sort of surrogate marker for overall survival.
Finding an early biomarker that is a surrogate for overall survival
One definition of a surrogate marker is: “any laboratory measurement or physical sign that is used in therapeutic trials as a substitute for a clinically meaningful endpoint that is a direct measure of how a patient feels, functions or survives and is expected to predict the effect of therapy.” It’s not just related, but it has to predict the effect of therapy as well.
Correlation by itself does not imply surrogacy; the figure (below) shows an example. The horizontal axis is the outcome in terms of the surrogate marker and the vertical axis is the true endpoint. There are two values, one for the control group (group C) and one for the experimental group (group E). In this example there’s a perfect correlation, so once you know what the outcome is in terms of the control group and the surrogate marker, you can tell exactly what the true endpoint value will be. The same thing applies for the experimental group: if you were given the outcome in terms of the surrogate marker for the experimental group you would know exactly what the true endpoint value would be, for example with median progression-free survival and median overall survival.
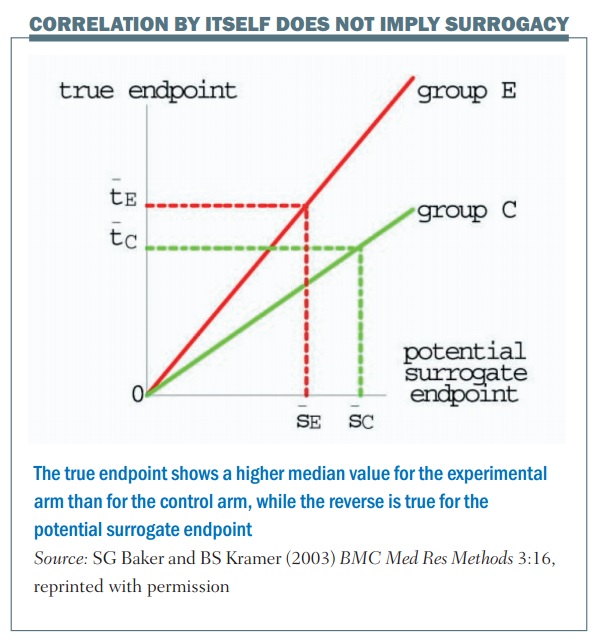
There is a perfect correlation in this example, but the problem is that this is not a good surrogate. This is shown by the dotted lines, which illustrate a larger value for the median surrogate endpoint in terms of the control group, giving a lower value for the true endpoint, if you compare between the control and the experimental group. What this illustrates is that, even though there is a perfect correlation between the surrogate and the endpoint for each particular value, it’s not a good surrogate endpoint.
From a statistical point of view we have to use specific criteria to define a surrogate. The most commonly used and gold standard criteria are the Prentice criteria defined in 1989 (RL Prentice, Stat in Med 1989, 8:431). This is defined as: “A test of the null hypothesis (H0) of no effect on the surrogate is equivalent to a test of H0 of no effect of treatment on the true endpoint.”
What does that mean? Essentially what we’re saying is that a marker can be used as a surrogate if it meets two conditions:
1. It predicts the final true endpoint
2. It fully captures the effect of the treatment upon the final endpoint.
This means we are looking at two different things, not just that the surrogate is related to the endpoint itself, but that it also captures the treatment effect.
Statistically, there are a couple of problems with the Prentice criteria. First, and most problematic, it is impossible to prove this condition, because it is saying that we have to prove a null hypothesis is true, and from a statistical point of view you can never prove that a null hypothesis is true. This means that we can’t follow the Prentice criteria strictly, though we can use them as a framework and relax the criteria slightly, and that’s what people have done in terms of trying to validate a statistical marker.
How do we do that from a statistical point of view? We have to demonstrate that there is a good correlation between the surrogate and the true marker. We also have to demonstrate that a good correlation exists between the treatment effects, so whatever the treatment effect is for the surrogate this has to be related to the overall treatment effect or indeed the true point of interest. We have to repeatedly demonstrate this both at the individual patient trial level and the individual trial level.
Validating a surrogate marker
An example of the statistical validation of a biomarker is a study of 5-fluorouracil-based therapy in colorectal cancer published in 2005 (JCO 2005; 23:8064–70). The research group used three-year DFS as a surrogate for the true endpoint of five-year OS. It required nearly 21,000 patients and 18 trials for the group to carry out this validation. There are three key plots in the results:
- The first plot (top left) looks at the relationship between three-year DFS and five-year OS. It shows the correlation is quite strong, with an R² value of 0.85. This means the three-year DFS is highly correlated with the five-year OS.
- The treatment effect plot (top right) looks at the hazard ratio between treatment arms in terms of DFS and OS. Again, there is a high correlation value of R², at 0.90. This mean that if you know the hazard ratio for DFS – the effect of treatment on the surrogate marker – you have a strong correlation with the hazard ratio for OS, i.e. the effect of treatment on OS.
- The third is a calibration plot (bottom graph) – if you have a hazard ratio for DFS, how well can you predict what the OS value would be? The graph shows that the OS hazard ratio is within about 95% of the predicted confidence intervals, as would be expected. As a result, we can conclude that the DFS hazard ratio can predict the OS hazard ratio reasonably well.
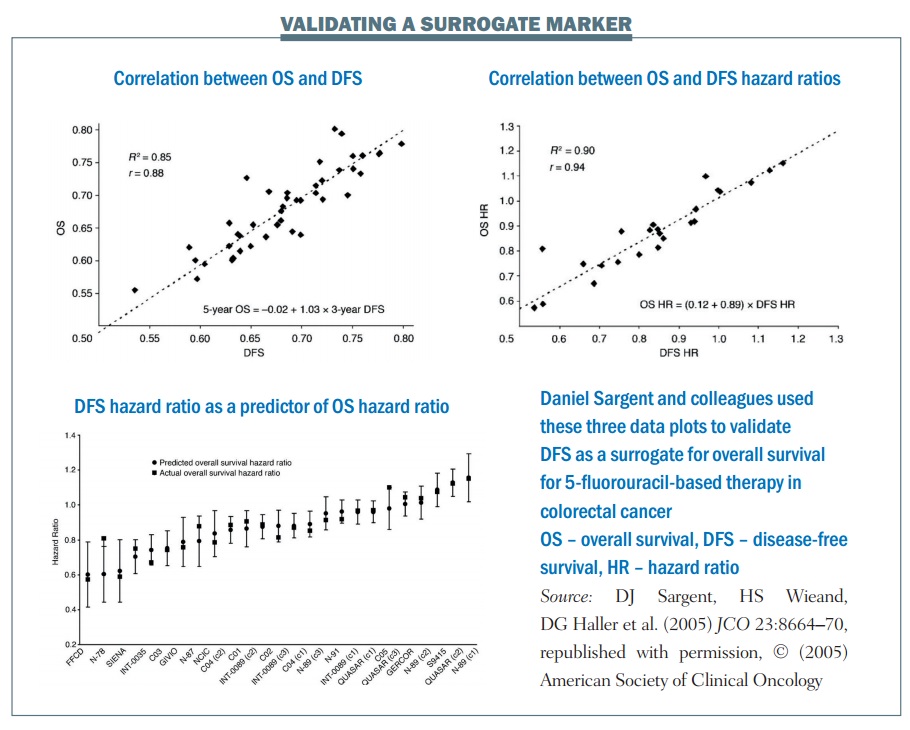
The group showed excellent correlation between the estimates of the surrogate marker and the true marker, or true OS. There was an excellent correlation between the treatment effects in terms of the hazard ratios and they showed an excellent calibration plot. There is also biological plausibility, in terms of DFS being related to OS. Finally, multiple formal analytical approaches were used to validate this, proving a validated surrogate marker. To quote Daniel Sargent, “These results suggest that DFS after 3 years of median follow-up is an appropriate endpoint for adjuvant colon cancer clinical trials of fluorouracil-based (5FU) regimens, although marginally significant DFS improvements may not translate into significant OS benefits.”
Do we need a surrogate in this context?
One question raised is whether we need a surrogate in this particular context: adjuvant colon cancer clinical trials of 5-fluorouracil-based (5FU) regimens? The use of 5-FU-based chemotherapy is going to be reduced as we move into a new era of molecularly targeted therapy.
This highlights one of the problems in statistical validations of a biomarker. What we have to do in clinical trials is to gather data to validate whether a surrogate is valid or not. But once we have those results, the surrogate may or may not be needed, as we already know the answers for that particular trial. This raises a bit of a difficulty with the validation of any particular marker.
Can DFS or PFS be used as surrogates for all clinical trials?
The next question is: can we use DFS, or in some cases PFS, globally for all clinical trials? Unfortunately, we cannot. In some settings DFS has become accepted as a surrogate, but it is not universal for every treatment in every single cancer.
What are the settings in which we can use these surrogates? There has been a lot of work into whether we can use DFS in particular settings, but we haven’t looked at every setting, because there are a lot of issues when trying to validate a surrogate. Generally, it has been recommended that we need 10 or more clinical trials to assess whether a marker is a valid surrogate, and it has to be validated every time for a specific treatment in a specific setting at a specific time point.
For example, in later work, Sargent et al note, “It is unlikely that the surrogacy of PFS for OS would have been demonstrated in the current context … with current salvage therapies.” What might prove to be a validated surrogate at one point may no longer be once there are more advanced second-, third- and fourth-line treatments.
Pragmatic validation
As researchers we are not really interested in what’s happened previously. We want a validated surrogate to use in future clinical trials. So how do we go about deciding whether or not we can use PFS or DFS as a surrogate marker for OS in future clinical trials? We have to settle for pragmatic validation, which means a biomarker has to:
- Have biological plausibility
- Have clinical utility demonstrated in clinical trials, for example having been validated in previous settings similar to the clinical trial being planned
- Satisfy clinicians, regulators, statisticians, and other researchers.
Early markers have the greatest potential benefit but are also the most difficult to validate because they are furthest away from when the true OS outcome occurs.
An ideal marker for a future clinical trial must be reliable, consistent, unbiased and clinically relevant. So is PFS/DFS an ideal marker? A study published several years ago (JCO 2009, 27:5965) looked at all the definitions used for different outcomes in clinical trials. Depending on the particular trial, DFS was defined in many different ways statistically, but the way the same definitions were used was not consistent from trial to trial.
Another issue that comes up when using PFS is when the timing of the evaluation of an event is not consistent between different treatment arms. This can make it seem as if progression is happening earlier in one arm than another, when in reality it is simply being recorded earlier.
A third issue is differential censoring. Patients do not necessarily leave a clinical trial just because of progression. Some stop the trial when they have adverse events and others may just decide to withdraw. These patients will generally be censored for the outcome of progression or PFS. But problems arise when the censoring itself is related to the outcome. For example, if a patient with grade 2 fatigue on a trial treatment believes it is working and if they have shown a small reduction or stabilisation of their disease they might be willing to tolerate the treatment a little bit longer. In contrast, a patient with the same grade 2 fatigue as an adverse event who does not believe the treatment is working may see a slight increase in their scan and come off treatment a little bit early. In this case, censoring is definitely related to the outcome. The problem is that this informative censoring may have a large effect on the outcomes, particularly if there is a different rate of informal censoring between treatments.
Summing up
In summary, PFS and DFS may often be poor surrogates for OS. It is very difficult to validate surrogate markers, although there is a lot of research trying to validate PFS and DFS in specific contexts. Unfortunately, validation often occurs too late to benefit particular clinical research, but it can be used as a basis for suggesting PFS and DFS may be useful for future studies.
The clinical relevance of PFS is unclear. As an independent outcome, PFS/DFS is most clinically relevant when there is the smallest benefit in clinical trials in terms of gain as a potential surrogate (that is, when PFS/DFS is most strongly related to OS, and the time from PFS/DFS to OS is small). Conversely, PFS/DFS would be most beneficial in clinical trials as a surrogate when in fact it has least clinical utility.
The use of PFS/DFS as a primary outcome in clinical trials is likely to increase, but it should be used with caution and understanding of all of the issues that affects its validity as a surrogate marker for overall survival.