



Testing for hereditary mutations that increase the risk of cancer is important for risk reduction, early detection and choice of treatment. Ephrat Levy-Lahad offers an overview of what we know ‒ and what remains uncertain ‒ about the rationale for testing, the risk implications, and how to discuss these with patients and families to enable them to make informed decisions.
This grandround was first presented by Ephrat Levy-Lahad, from the Shaaare Zedek Medical Center, Jerusalem, as a live webcast for the European School of Oncology. It was edited by Susan Mayor. The webcast of this and other e-sessions can be accessed at e-eso.net
Germline testing is carried out first for the cancer patient and second for the patient’s relatives. For the patient, testing has implications for treatment. One example is the extent of surgery. If breast cancer occurred because of a genetic risk, the patient might elect to have bilateral mastectomy even if lumpectomy was sufficient for management of the cancer itself. For colon cancer, if there is a genetic predisposition, the surgeon might choose to perform a wider excision compared with non-hereditary colon cancer.
Non-surgical treatments may also be tailored, e.g. PARP inhibitors for ovarian cancer in BRCA1/BRCA2/Fanconi pathway carriers, or avoidance of radiotherapy in TP53 carriers. Beyond treatment of the cancer itself, if the patient has an inherited predisposition they may be at risk for additional tumours, and these also need special surveillance and treatment. For example, a BRCA1 carrier with breast cancer is also at risk for ovarian cancer, and a female patient with Lynch cancer syndrome is also at risk for endometrial cancer.
The other important people in this equation are the relatives. Once we find a mutation in a patient, we can test the relatives to find out whether or not they inherited the mutation. Non-carriers in the family generally have background risk and do not require special surveillance or prevention options. Carriers have increased risk and should be given the opportunity of special surveillance and prevention.
What are gene panels?
Gene panels are essentially tests based on Next Generation Sequen-cing (NGS), which can test multiple genes simultaneously. There are two main types of panel:
- Tumour-, organ- or syndrome-specific, such as a colon cancer panel or the hereditary breast/ovarian panel,
- Pan-cancer panels that include all of the known hereditary predisposition genes. These include many more genes than the tumour- or syndrome-specific panels.
We can generally distinguish three types of gene on panels:
- Established hereditary cancer genes known to cause specific cancer syndromes. Examples include APC in colon cancer, BRCA1/2 for hereditary breast/ovarian cancer and VHL for Von Hippel-Lindau renal cancers,
- Genes more recently identified as having strong evidence for being cancer predisposition genes, e.g. RAD51C for hereditary breast/ovarian cancer and GREM1 for colon cancer.
In the third category, which is more problematic, are genes with lesser evidence where the risk for specific cancers is unclear.
Generally speaking, there is a core list of genes included in practically all panels. These include the established hereditary cancer syndrome genes and those with strong evidence of association with specific cancer risks. However, genes with lesser evidence are more variable between panels, and there is no single consensus list of genes that are found on all panels.
Why are some genes with less evidence included on gene testing panels?
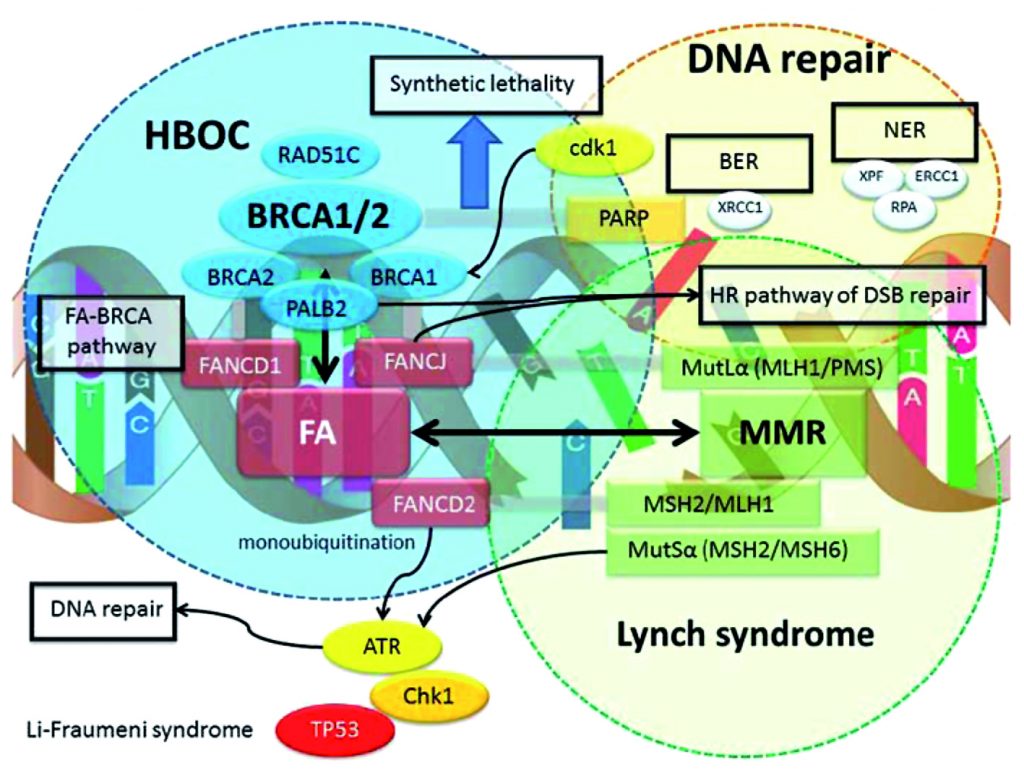
Some genes are included in panels because of ‘guilt by association’. Over the last few decades it has become very clear that mutations in DNA repair genes are common causes of hereditary cancer predisposition. For example mutations in genes involved in mismatch repair such as MLH1 and MSH2 cause Lynch syndrome. BRCA1/2 and PALB2 are all part of the Fanconi anaemia pathway, which is important for homologous recombination DNA repair.
The involvement of DNA repair mutations in inherited cancer predisposition is quite logical because defects in DNA repair lead to mutation accumulation, and this is thought to lead to tumourogenesis (see figure). However, the fact that a particular gene is part of a specific DNA repair pathway does not necessarily mean that mutations in this gene will be associated with a specific risk. CHK1 or ATR are often included in panels, although it is, as yet, unclear whether they are associated with predisposition to cancer, and if so, for which specific cancers.
In general, we can distinguish high-risk or high-penetrance genes versus those that are associated with moderate or low risk. High-risk genes are generally associated with a relative risk for a particular cancer that his more than four times the risk in the general population. Moderate-risk genes confer a relative risk of between two and four times that in the general population. Low-risk genes have a relative risk of less than two.
There are also specific variants that can be associated with different levels of risk. For example, although APC and BRCA2 are both very-high-risk genes, there are specific mutations that are associated with low risk, such as the I1307K mutation in APC and the polymorphic stop p.K3326X mutation in BRCA2. Finally, there are genes without any evidence-based risk, and so there are no guidelines on how to treat patients with mutations in these genes.
How to act on the results of a gene panel
Technically speaking, the result of a gene panel is the identification of a variant. The American College of Medical Genetics introduced a five-category – or five-tier – system that is now commonly used. Variants can be ‘pathogenic’ or ‘likely pathogenic’, in which case they are reported. They can be ‘likely benign’ or ‘benign’, in which case they are not reported. In the middle, there is a ‘black box’ of variants of unknown significance (VUS), and whether or not these are reported is a matter of lab policy. Some labs report VUS and some do not.
Identification of a variant is the technical result. However, as clinicians we would like to have a ‘bottom line’, with a result that is either positive, indeterminate or unclear, or negative:
- A positive result is when a pathogenic or likely pathogenic mutation is identified in a high- or moderate-risk gene.
- An indeterminate result is the identification of a variant of unknown significance in a gene known to be important, or any variant in a gene with unclear significance.
- A negative result can be a true negative in the sense that the patient has no inherited predisposition. However, a negative result depends on the state of current knowledge regarding the particular genes tested. A patient with a very young age of onset or significant family history might have a genetic predisposition for their cancer, even though the particular gene has not yet been identified.
What are the pros and cons of panel testing?
The main advantages of panel testing are that it is fast and provides simultaneous testing of multiple genes. This is important, particularly if there are many genes that can cause a particular cancer or cancer syndrome. It is also helpful in time-sensitive situations, such as when a quick decision is needed on the surgical approach.
Testing gene by gene risks patients being lost to follow-up, while simultaneous testing for several genes means fewer patients will not complete testing. The cost is much cheaper, with the cost of a panel being about the same as classical sequencing of a single gene. Panels are also less syndrome- specific, which means clinicians are less dependent on family history.
The cons of panel testing are mainly related to the fact that we can test for a lot more than we can understand, limiting interpretation of results. Some genes included in panels have limited evidence, and no guidance for clinical action.
Variations of unknown significance occur at a frequency of 10–40%, depending on the lab policy for reporting and the number of genes tested. The larger the panel, the more genes are first tested and the greater the chance of finding a variation of unknown significance.
This has significant unwanted outcomes, and often leads to overtreatment or over-screening because it is very difficult to ignore a variant once it has been identified.
An additional problem is the issue of quasi-incidental cancer mutations, which means finding a pathogenic mutation in a gene that is not related to the patient’s cancer. For example, finding a Lynch syndrome mutation in a breast cancer patient may mean the Lynch mutation caused the breast cancer, but this is often unclear.
Yield of panel testing for specific cancers
Breast cancer
Breast cancer has been the most extensively studied cancer with regard to the yield of testing panels. In patients who have already tested negative for BRCA1/BRCA2 mutations, the chance of identifying a different mutation in another gene is around 5%. In a patient who has had no genetic testing, panels including BRCA1/BRCA2 will have a total yield of around 15%: 10% will be BRCA1 or BRCA2 mutations, and about 5% will be in other genes, mostly moderate-risk genes such as ATM and CHEK2. Overall, BRCA1/BRCA2 account for most of the currently identifiable high-risk genetic predisposition for breast cancer.
Ovarian cancer
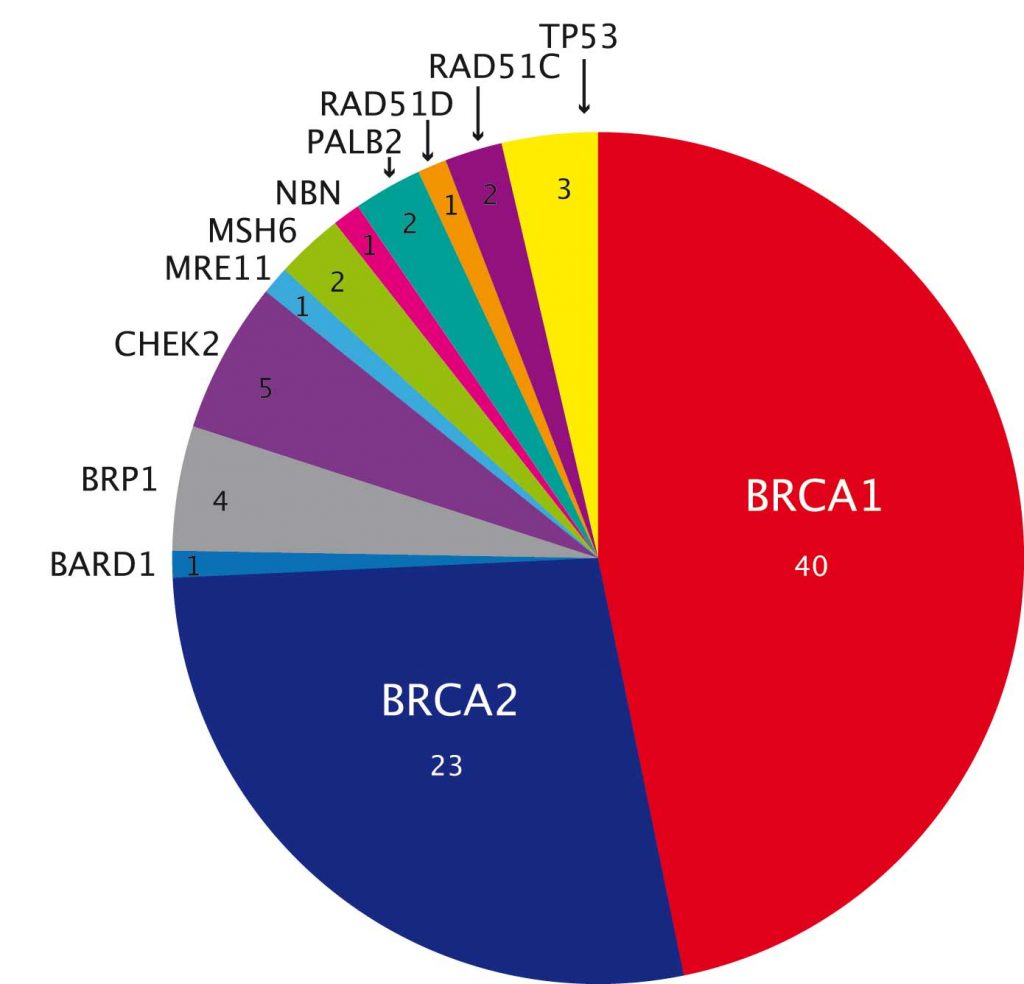
A study in ovarian cancer showed significantly higher overall yield, at over 20% (PNAS 2011, 108:18032–37). More than two-thirds are mutations in BRCA1 or BRCA2, but there are also mutations in genes such as TP53 and CHEK2 (see figure above), which are not clearly linked to ovarian cancer.
One in three patients with mutations were diagnosed after the age of 60. More testing in larger numbers of individuals shows that even individuals with little family history and those of older age can have inherited mutations.
Colon cancer
The yield of panel testing for colon cancer is about 10–15%. The distribution includes more high-risk genes and fewer moderate-risk genes compared to breast cancer, with a lower predominance of particular genes and greater heterogeneity of the genes involved (Annu Rev Genom Hum Genet 2017, 18:201–27).
There may be more clearly pathogenic results in colon cancer, with fewer variations of unknown significance (Gastroenterology 2015, 149: 604–13). However, this could be a result of patient selection, as many tumours today are tested directly for mismatch repair deficiency either by microsatellite instability or immunohistochemistry for MMR proteins, such as MSH2, MSH6 or MLH1.
Perhaps panel testing is more likely in patients who have already been shown to have MMR deficiency and are, therefore, more likely to harbour inherited mutations.
Pancreatic cancer
In pancreatic cancer a recent study found that 3.5% of patients were carriers of known genes (JCO 2017, 35:3382–90), including 0.4% carriers of mutations in candidate genes.
Mutation distribution was somewhat different from other cancers, with BRCA2 and ATM accounting for almost 70% of mutations; BRCA1 was a much more minor player, as were PALB2 and MLH1. The important message regarding pancreatic cancer is that the variants identified, although rare, are targetable. This has clear therapeutic implications for utilising PARP inhibitors (BRCA1 and BRCA2, ATM, PALB2) or immune checkpoint inhibitors (MLH1).
Prostate cancer
A 20-gene panel of DNA repair genes identified deleterious variants in 11.8% of men with metastatic prostate cancer (NEJM 2016, 375:443–53). This was much more common than the rate of 4.6% seen in men with local prostate cancer, and the 2.2% rate found in population controls from the Exome Aggregate Consortium, which assesses the frequency of variants in tens of thousands of individuals.
This is an important point, because many previous studies have only collected data on the frequency of variants in patients with cancer, without comparing their frequency to that in the general population.
The prostate cancer study also showed that age at diagnosis and family history did not significantly affect yield. The mutation distribution of prostate cancer was somewhat reminiscent of that for pancreas cancer. The major culprit was BRCA2, accounting for 44% of mutations; next was ATM, with 13%. CHEK2 and BRCA1 were more minor players.
Again, this has therapeutic implications, because olaparib, the oral PARP inhibitor, has been approved by the FDA as a monotherapy for previously treated, metastatic, castration-resistant prostate cancer for people with BRCA1/2 or ATM mutations.
Renal cancer
Considering renal cancer as an example of a less common cancer, a 19-gene panel found that 6.1% of all renal cancer patients had a mutation (Cancer 2017, 123:4363–71). The most common mutations were in the FLCN gene (1.8%), which causes a cancer syndrome known as Birt-Hogg-Dubé.
Fumarate hydratase mutations occurred in 1.3% of patients, and the mutation rate in VHL, which is a canonical renal cancer gene, was only 0.2%. This could be a result of prior selection, i.e. if patients with a clear history suggestive of Von Hippel-Lindau had single-gene testing, and thus those found to have VHL mutations were not tested using panels.
There was a high rate of variations of unknown significance (18.4%), often in large genes or genes that have pseudogenes (genomic DNA sequences similar to normal genes but non-functional) that complicate testing, such as TSC2 (tuberosclerosis 2 gene), MET and PMS2.
Pan-cancer panels
A 76-gene panel tested in 1,040 patients (median age 58 years) with advanced cancer showed that 17.5% had a clinically actionable mutation (JAMA 2017, 318:825–35); 14% had a moderate- or high-penetrance mutation. Half of these would not have been detected based on their family history, age or tumour type. Only about 4% were actionable for targeted therapy in the patients.
Regarding the distribution of mutations in this study, about 40% of mutations were either in BRCA1 or BRCA2. BRCA1 was more specific for breast and ovarian cancer, but BRCA2 was associated with a much wider spectrum of different cancers.
Resources to help manage patients with a reported variant
ClinVar (short for clinical variation) is provided as a general resource by the US National Center for Biotechnology Information. It enables searches by gene and by variants within genes, showing how the variant has been classified and the evidence for the classification.
It is becoming increasingly useful for understanding variants as more information is added. There are also gene-specific databases, including for BRCA1, BRCA2 and TP53, and databases for Lynch syndrome and other hereditary cancers.
A number of guidelines provide information on care and follow up for patients with pathogenic or likely pathogenic variants in specific genes, in addition to prevention and surveillance guidance for relatives who are known to be carriers.
For example, the US National Comprehensive Cancer Network (NCCN) guidelines are updated annually and include recommendations regarding multiple genes, including genes for hereditary breast and ovarian cancer (including ATM, BRCA1/2, BRIP1, CDH1 etc) and for colon cancer (including APC, with a separate recommendation for the I1307K mutation, BMPR1A etc).
Genetic counselling for panel testing
Genetic counselling has traditionally been given both before testing, to allow the patient an informed decision about whether to be tested, and after testing, when the results are available.
The patient should be made aware of all the ramifications of testing before they make a decision to be tested. This tends to be less of a concern for cancer patients, who are often very interested in testing, as it could impact their treatment.
Pre-test counselling
Pre-test counselling should include a discussion of the concept of inherited cancer risk and a detailed review of the pedigree, including ethnic background, overall family structure, age(s) at diagnosis and type(s) of cancer in affected family members.
It should provide information on gene mutations of interest and explain that different mutations have different cancer risk. It is not realistic to detail risks for every gene tested, but the aim should be to give an idea that some mutations are high risk, while others lead to moderate or low risk, or are of unknown risk.
Pre-test counselling should explain options and limitations of surveillance and prevention. Specific mention should be made of high-penetrance syndromes with impactful management strategies, such as CDH1 mutations and prophylactic gastrectomy.
Patients need to know that they might be offered quite aggressive measures. We should discuss the possibility of getting uncertain results and variations of unknown significance. The implications for other family members should also be discussed.
The issues of cost and insurance coverage should be covered in countries where genetic results can influence ability to be insured. Confidentiality issues should be noted and it is essential to discuss how the patient would like to receive their results.
In terms of informed consent, a test should ideally offer the option to opt out of receiving information on variations of unknown significance, because often neither the physician nor the patient knows what to do with this result. Patients should also be offered the option not to receive information on genes that are unrelated to their cancer. However, many labs do not offer these options.
Post-test counselling
Post-test counselling has two major components: genetic issues and medical follow-up. The genetic element involves explaining the test results and the qualitative and quantitative cancer risks.
If a pathogenic or likely pathogenic mutation is found, there needs to be a discussion of the inheritance, which relatives should be tested, and how to contact relatives. If no mutation is found, there should be a discussion about whether there is still suspicion that there might be a genetic syndrome.
With regards to medical management, any early detection or risk reduction strategies should be discussed and types of therapy that might be available should be explored. There should also be a discussion of clinical trials, registries and recommendations for follow-up.
Take home messages
- Gene panels are a major advance in genetic testing, offering un-biased analysis of inherited predisposition in a timely manner and at reasonable cost.
- Panels should be chosen based on the patient’s characteristics, their family history, the genes in the panel, the reporting policy on variations of unknown significance, and previous genetic testing.
- Panel yield is 5–15%, depending on the tumour type and previous genetic testing.
- Actionable outcomes are not very common, but when they occur they are important and include targeted therapy, specific surveillance and prevention, and testing of family members.
Panel yield: overview
- Current studies of panels indicate a 5–15% yield overall, depending on cancer type, but some studies have detected higher rates
- Rates of variations of unknown significance are between 10% and 40%.
- Most studies do not compare mutation rates in patients against controls.
- Figures are likely to be overestimates due to ascertainment bias, because people participating in studies generally have younger than average age of onset and are often more severe cases.
- Gene distribution may be biased by previous single-gene or family-based testing.

