Could tailoring treatments to broad taxonomies work where targeting individual – or even multiple – genetic mutations has not? A growing number of researchers working on specific tumour types and/or across tumours believe this integrative approach, involving ‘precision classification’, could be the way to go. Janet Fricker talked to some of the key players.

In a prescient Cancer World guest editorial published in 2005, Alberto Costa, breast surgeon and head of the European School of Oncology, wrote, “The whole concept of breast cancer as a single disease is now dead, and we therefore need to make fundamental changes in the way we approach treatment decisions.”
The editorial was a response to the 2005 St Gallen conference, which had concluded that breast cancer should be characterised according to eight elements: size, histological type, grading, hormone receptor status, lymph node status, proliferation index (ki67), cErbB2 status, and the presence or absence of peritumour vascular invasion.
In 2018, routine clinical assessment of breast cancer still comprises morphological assessment (size, grade, lymph node status), and testing for oestrogen and progesterone receptors (ER and PR) and HER2. Such information allows pathologists to classify breast cancer into four subtypes: luminal A cancers (usually ER+ and/or PR+ with a low proliferation index); luminal B cancers (ER+ and/or PR+ and high proliferation index); HER2-amplified cancers (can be either ER/PR positive or negative, but with high levels of HER2); and basal-like tumours (which are ‘triple negative’, i.e. negative for ER, PR and HER2).
However, it is now widely recognised that this grouping does not reliably predict how tumours behave.
“From our clinic experience we realised that breast cancer patients have very disparate outcomes and that it is a misnomer to call it a single disease, or even one with four subtypes,” says Carlos Caldas, who in 2012 published a landmark study demonstrating that breast cancer is an ‘umbrella term’ for at least 10 separate diseases (Nature 2012, 486:346–52).
This new breast cancer stratification was validated in a subsequent paper by the Caldas group (Genome Biology 2014, 15:431).
“Personalised medicine is about good taxonomy. When treating bacterial infections you need good classification to know whether you are treating gram-positive or gram-negative infections. In much the same way, for effective treatment of cancer you need proper molecular stratification of tumours,” says Caldas, from Cancer Research UK’s Cambridge Institute.
A revolution in tumour pathology
In the intervening years the METABRIC project, a joint project between Caldas’ group and Sam Aparicio’s group at the University of British Columbia, has spurred a revolution in breast cancer stratification. The collaboration has been largely responsible for moving tumour classification beyond examining tissue under a microscope to pinpoint abnormal anatomy, to a system that incorporates extensive molecular profiling.
“Ultimately we hope that our ‘iCluster’ approach will help doctors treat diseases better based on specific genetic signatures”
In METABRIC (see The Molecular Taxonomy of Breast Cancer box), investigators used microarrays to delve into the DNA and RNA of tumours. They also tested each tumour sample for alterations in copy number, because copy number aberrations were known to dominate the breast cancer genomic landscape.
The resultant large-scale, multi-dimensional dataset, which incorporated samples from 2,000 women with breast cancer, together with data on their clinical outcomes, was navigated using novel high-performance computational and statistical techniques.
In an epic effort, the investigators sifted through gigabytes of information to extract meaningful patterns in an analytical approach known as ‘data mining’.
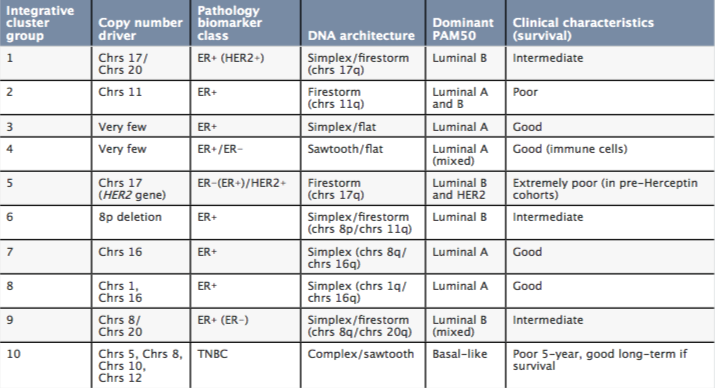
The result was 10 integrative clusters, or ‘iClusters’ (see The Molecular Taxonomy of Breast Cancer table), which were later expanded to 11 clusters, after cluster 4 was further subdivided into tumours that were ER positive and negative (Nature Communications 2016, 7:11479).
“The basic tenet of medical practice is that the better you phenotype a disease, the more likely you are to treat it correctly,” says Caldas, who initially trained at the University of Lisbon.
“Ultimately we hope that our ‘iCluster’ approach will help doctors treat diseases better based on specific genetic signatures.”
The iCluster methodology, which has become known as ‘integrative medicine’ or ‘precision categorisation’, has since been utilised to explore a range of cancers, including:
- Prostate: divided into five subtypes by the CamCaP project (EBioMedicine 2015, 2:1133–44),
- Pancreatic: divided into four subtypes by Andrew Biankin (Nature 2015, 518:495–501),
- Colorectal: divided into four groups by Angurah Sadababdam (Nature Medicine 2015, 21:1350–56),
- Bladder: divided into five subtypes by Seth Paul Lerner (Cell 2017, 171:540–556.e25),
- Melanoma: divided into four subtypes by researchers from The Cancer Genome Atlas Network (Cell 2015, 161:1681–96).
While the groups stratifying each of these cancer types all took broadly similar approaches, they analysed different combinations of data sets, including DNA and RNA, single point mutations, copy number, whole genomes and other properties of tumours.
Breast cancer integrative clusters (iClusters)
In a study looking at the somatic mutation profiles of breast cancers, Carlos Caldas sequenced 173 genes in samples taken from almost 2,500 patients with breast cancer, and showed that PIK3CA (coding mutations in 40.1% of the samples) and TP53 (35.4%) dominated the mutation landscape (Nature Communications 2016, 7:11479).
Only five other genes harboured coding mutations in at least 10% of the samples: MUC16 (16.8%); AHNAK2 (16.2%); SYNE1 (12.0%); KMT2C – also known as MLL3 – (11.4%) and GATA3 (11.1%).
These word clouds illustrate the distributions of mutations in the 173 sequenced genes in four integrative clusters, with the size of each word corresponding to the relative frequency of the mutations observed for a given gene in each cluster.
Making sense of complexity
Initiatives like these are helping investigators to ‘gain a handle’ on the ecosystems involved in growth of tumours, and to start to acquire more of a holistic understanding of the complexity of cancer, by including information about a wider group of genes, says Caldas, who sees it as a pragmatic approach to dealing with massive complexity. “The idea that every tumour is different from all others represents an impossible task. Subdividing cancers into different subtypes provides the closest approximation that we can get to the truth,” he says.
Andrew Biankin, from University of Glasgow, who now chairs the International Cancer Genome Consortium, agrees: “The integrative approach allows cancers to be broken down into manageable subtypes that help us to understand similarities and then design drugs against shared mechanisms.”
Caldas stresses that the characterisation of breast cancer into subtypes has yet to affect the way patients are managed. However, he firmly believes that the 11 subtypes offer the eventual possibility of a platform to investigate new treatments.
“At the moment trials are more about the drug than the disease. Hopefully studies like METABRIC offer the possibility to change that and start to tailor treatments to the disease,” says Caldas.
“The clusters in effect provide a grouping of biomarkers that can be used to test new treatments”
In a recent paper (Nature Communications 2016, 7:11479), Caldas and colleagues investigated the frequency of 173 genetic mutations across 2,500 breast cancer patients, and showed that patients in the same iCluster demonstrated similar patterns of mutations (see above). Since some of these genes are known to be involved in the production of enzymes within human cells, they could provide targets for the development of new anti-cancer drugs.
Timeline – Exploring the Cancer Genome
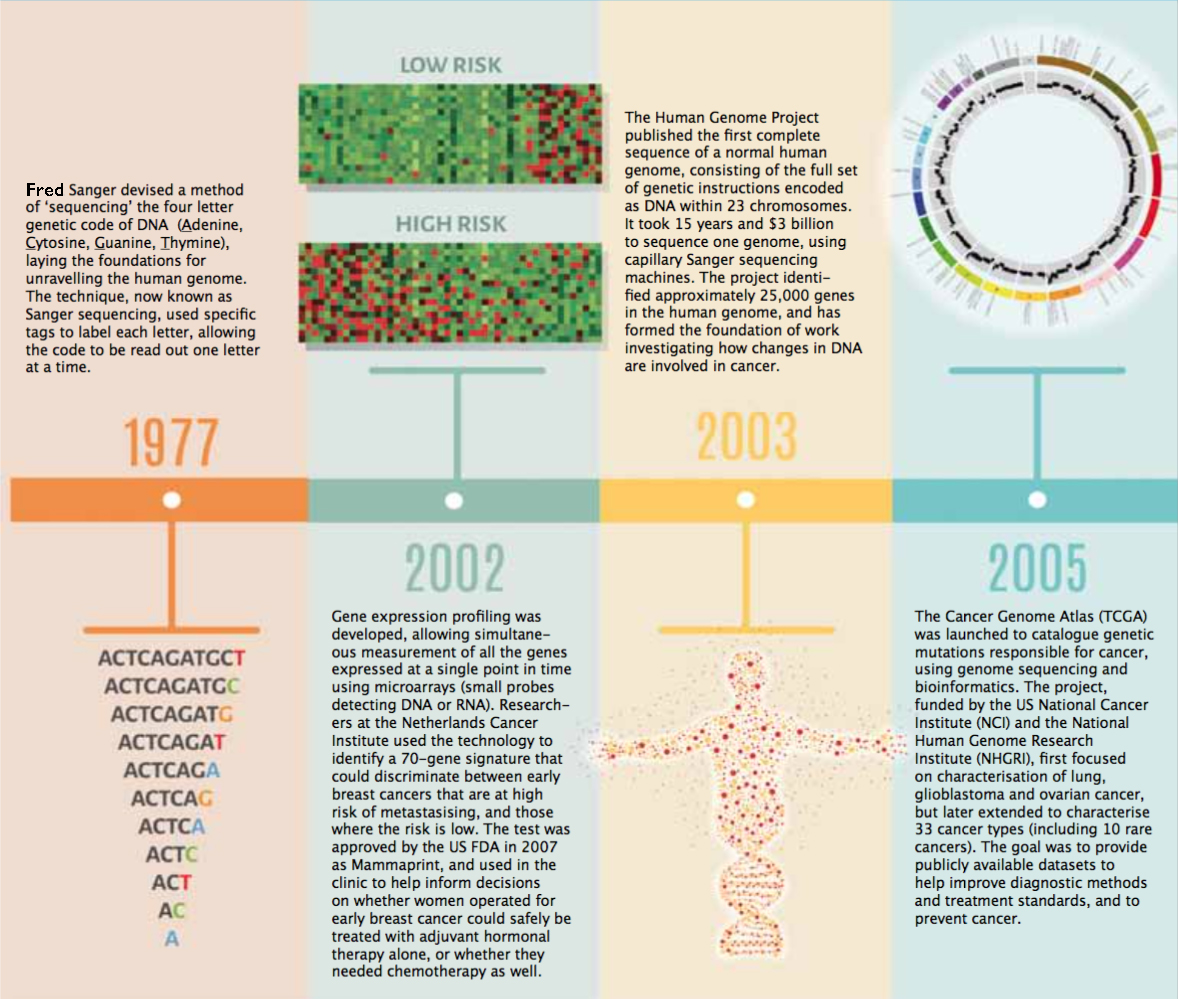
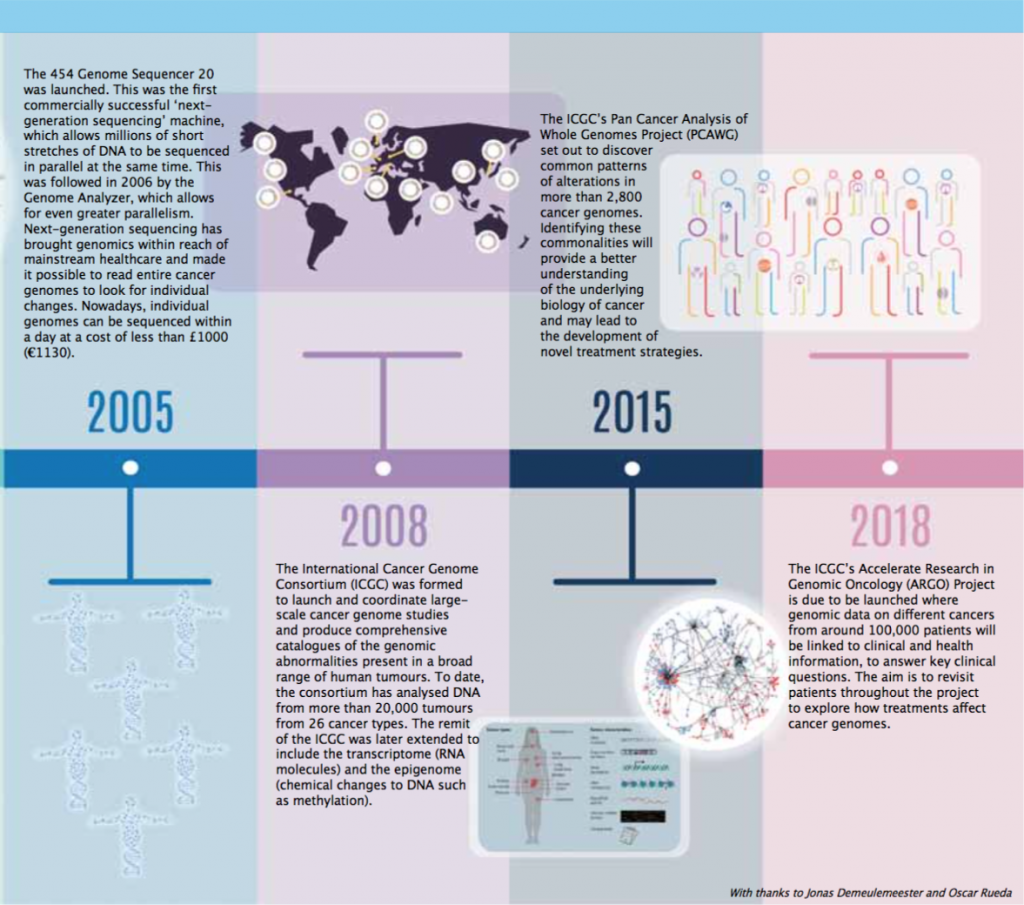
A guide to diagnosis, prognosis and hopefully treatment
“The clusters in effect provide a grouping of biomarkers that can be used to test new treatments,” said Heinz Zwierzina, from Innsbruck Medical University, who chairs the Cancer Drug Development Forum.
Caldas’ next goal is to devise a simple molecular test that could be performed on routinely collected paraffin block samples, to prospectively assign patients to one of the 11 subtypes. Once patients have been characterised into the different subtypes it would then be possible to follow these subgroups in clinical trials to explore which agents work best for each.
To accelerate the drug testing process, Caldas and his team have developed a technique where human breast cells grown in mice can be removed to run further tests using experimental drugs in vitro (Cell 2016, 167: 260–74). The approach, says Caldas, reflects the biological reality of cancer more accurately than growing cells in plastic dishes, which is known to differ from the way cells grow inside the body. “Testing all the new treatments on patients with the 11 different breast cancer subtypes would take centuries and tens of thousands of patients. We hope this approach will help speed things up,” he explains.
In addition to helping drug development, the integrative approach can be used to provide prognostic insights for patients. In breast cancer, for example, Caldas’ team have found that 40% of patients with breast cancer classified as cluster 2 or cluster 5 are alive 15 years after diagnosis, while 75% of those with cancers classified as cluster 3 or cluster 4 are alive at the same time point.
The same approach can also be used to identify the groups that would benefit from other treatment approaches, including surgery, radiotherapy and active surveillance. “In prostate cancer, molecular signatures associated with the most aggressive disease could be used to provide a rationale for early adjuvant treatment immediately after prostatectomy or for undertaking active surveillance,” says Alastair Lamb, a prostate cancer surgeon from Oxford, who led the CamCaP project while training in Cambridge.
Integrative data can also help diagnosis, providing investigators with additional ‘flags’ to look for in liquid biopsies – an approach that uses tumour DNA shed into the blood to track cancers in real time. “This could change the way we monitor patients and may be especially important for people with cancers that are difficult to reach, as taking a biopsy can sometimes be quite an invasive procedure,” says Caldas.
Targeting mutations has been ‘a major disappointment’
The integrative approach contrasts to the ‘reductionist’ approach of cancer personalised medicine, where investigators have focused on treating one component of the tumour, such as an aberrant enzyme or protein. “The successes of imatinib in CML, crizotinib in NSCLC and trastuzumab in breast cancer gave the impression that targeting single molecular alterations was easy,” says Vassilis Golfinopoulos, Medical Director at EORTC, Europe’s largest cancer clinical trials organisation. “However, the reality is that these agents represent only a tiny percentage of targeted drugs, with many more having failed to show significant efficacy in clinical trials.”
Leif Ellisen, Program Director at the Massachusetts General Hospital Center for Breast Cancer and Professor of Medicine at Harvard Medical School, agrees, and says the reason why the approach of targeting a single gene or mutation has been so disappointing is because cancer has so many ways to subvert the effects of inhibiting one pathway.
He cites as an example the transitory impact BRAF inhibitors have in melanoma patients with the BRAF V6000 mutation, due to the ability of the cancer cells to get around the inhibited BRAF through activating the MAPK pathway.
Hopes of fixing the problem by targeting multiple pathways are largely failing in practice, he adds “because the toxicity is additive, with the result that combinations aren’t tolerated.”
“By exploring complex data we can identify potential common denominators that would not be so open to developing resistance”
“Taking into account the heterogeneity of cancer, it’s highly unlikely that many tumours would be regulated by a single driver,” says Jan Brábek, a cell biologist from the Charles University, Prague, with a special research interest in cancer cell invasiveness and metastasis. “It’s only by exploring complex data that we can hope to find patterns of drivers and identify potential common denominators that would not be so open to developing resistance.”
The new ‘integrative’ paradigm
To explain the potential of integrative medicine, Brábek uses the analogy of a ‘getaway’ car in a bank robbery. “If you shoot one of the drivers it’s all too easy for another to take over the wheel, which is in effect what happens with resistance. However, if you target more fundamental mechanisms, such as shooting the wheels, you can prevent the possibility of anyone else being able to take over. This enables you to stop the car completely.”
Possibilities for more fundamental agents that could be explored in the ‘iCluster’ subgroups, he suggests, could include anti-invasive and anti-metastatic agents and drugs targeting tumour metabolism.
Integrating new types of data into the taxonomy
New concepts and approaches to exploring the cancer genome are continually becoming available, which could further refine the iCluster classifications
One concept is that the tumours could in theory contain a number of different iClusters side by side. The evidence for this comes from Charles Swanton, now at the Francis Crick Institute, London, who analysed the entire genomes of seven individual samples taken from a single renal tumour, and found that only around one-third of more than a hundred separate mutations he identified were present in all samples (NEJM 2012, 366:883–92).
As point mutations and copy number aberrations tend to change over the course of the illness, account also needs to be taken of how cancer gene expression evolves with time and whether iCluster definitions might change.
Serena Nik-Zainal, from the Wellcome Sanger Institute, Cambridge, has been characterising patterns of mutations, known as ‘mutational signatures’, which include base substitutions, small insertions/deletions, rearrangements and copy number changes.
“Whole genome sequencing allows us to read every single mutation in a cancer genome, which includes not just ‘drivers’ but also passenger mutations as well,” says Nik-Zainal, who adds that, while passenger mutations may not have caused the initial cancer they can have significant effects on the biology of tumours.
In the first paper, Nik-Zainal explored the whole genome sequence of 21 breast cancers and created a catalogue of more than 200,000 different mutations that had occurred over the course of the patient’s life (Cell 2012, 149:994–1007).
In a second study of the genomes of 560 women with breast cancer, Nik-Zainal found five new genes associated with breast cancer and 13 new mutational signatures influencing tumour development. (Nature 2016, 534:47–54; Nature Communications 2016, 7:11383).
“New concepts and approaches to exploring the cancer genome are continually becoming available”
Nik-Zainal is now working with Caldas, Jean Abraham and others in the Personalised Breast Cancer Project, launched in Cambridge at the end of 2016, to combine the mutational signatures obtained from a highly detailed DNA profile of 2,250 breast cancer patients with the iCluster subgroup classifier (to date more than 200 patients have been recruited into this clinical molecular study). “In effect we are combining two integrative approaches to provide further integration, to see if we can split patients into yet smaller cohorts to better inform treatment decisions,” Nik-Zainal explains.
Unfazed by the prospect of future subdivisions making his 11 subgroups obsolete, Caldas draws comparisons to plant taxonomy. “Each subtype can be considered as a type of tree. One subgroup is composed of olive trees, another of pine trees, and another of beech trees. While all olive trees are not identical, the pattern of their branches, leaves and flowers are similar and very different from those of pine. We can be confident that the olive tree will not evolve into the pine tree,” he said.
The Molecular Taxonomy of Breast Cancer
PAM50 ‒ breast cancer molecular subtyping in current use; Chrs ‒ chromosome; ER ‒ oestrogen receptor; TNBC ‒ triple-negative breast carcinoma Source: HG Russnes, OC Lingjærde, AL Børresen-Dale, and C Caldas (2017) Am J Pathol 187: 2152‒62. © 2017. Reprinted with permission from Elsevier
The Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) undertook an integrative analysis of tissues samples from breast cancer patients that resulted in the landmark definition of breast cancer as a constellation of 10 genomic-driver-based subtypes (Nature 2012, 486:346‒52).
The project, representing the largest molecular profiling study ever undertaken, was led by Carlos Caldas, from Cambridge University, and Sam Aparicio, from the University of British Columbia, Canada.For the analysis, investigators obtained 1,000 frozen breast cancer samples from five tumour banks in the UK and Canada. DNA and RNA were isolated from samples and then hybridised to microarrays (state of the art for 2011), which had around two million probes for DNA, RNA and increased copy numbers.
This research was enabled by the biobank infrastructure in both Cambridge and Vancouver, which allowed tumour samples to be linked with detailed information about clinical outcomes and treatment of patients. Remarkably, every patient in METABRIC now has had a minimum of 10 years’ follow-up.Additionally, blood samples from 550 patients were available, allowing the group to compare tumour DNA with normal DNA in individual patients. For individuals with no matched ‘normal’, their tumour DNA was compared to an average of 500 ‘normals’. From this approach, the team were able to identify when a copy number was not a tumour aberration, because some people had this pattern in normal DNA.
Investigators used computer algorithms to search for patterns, or integrative clusters, based on similarities in copy number variants, single nucleotide polymorphisms and somatic copy number aberrations, SNPs, and gene expression, and whether they shared similar outcomes. Altogether, the team identified 10 groups of tumours (listed above) that behave consistently. The team went on to validate these grouping with a second cohort of 1,000 biobank breast cancer samples and a third cohort of 7,500 biobank breast samples (Genome Biol 2014, 15:431).
More recently, the team have subdivided the fourth group into whether patients are oestrogen receptor positive or negative, providing 11 subgroups (Nature Communications 2016, 7:11479). Changes in copy number led to the identification of 40 putative cancer driver genes, including PIK3CA.